| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://wjon.elmerpub.com |
Original Article
Volume 16, Number 5, October 2025, pages 471-477

Problems in Cancer Genome Medicine: Base Mutations Cause Intron Start Signals, Resulting in Unexpected Splicing
Takuma Hayashia, c, Ikuo Konishia, b
aCancer Medicine, National Hospital Organization Kyoto Medical Center, Kyoto-city,
Kyoto 612-8555, Japan
bDepartment of Obstetrics and Gynecology, Kyoto University
Hospital, Kyoto-city, Kyoto 606-8507, Japan
cCorresponding Author: Takuma
Hayashi, Cancer Medicine, National Hospital Organization Kyoto Medical Center, Mukaihatake-cho
1-1, Fukakusa, Fushimi-ku, Kyoto-city, Kyoto 612-8555, Japan
Manuscript submitted June 24, 2025, accepted August 14, 2025, published online September 17,
2025
Short title: Base Mutations Cause Unexpected Splicing
doi:
https://doi.org/10.14740/wjon2631
| Abstract | ▴Top |
Background: The genetic characteristics of surgically removed cancerous tissues are examined using cancer gene panel testing in cancer genome medicine to detect the pathogenic variants involved in the proliferation and progression of cancer cells. An antitumor drug is prescribed if it directly acts on the detected pathogenic variant; however, some aspects require careful consideration by medical professionals in such cases. The genetic mutations involved in the progression or onset of malignant tumors differ with race. Furthermore, genetic mutations that are variants of unknown significance (VUS) may be involved in the progression or onset of malignant tumors in some races according to the ClinVar results from the National Center for Biotechnology Information. Single nucleotide variations can result in silent mutations or splice sites.
Methods: We therefore reexamined the CGP results (VUS) of patients suspected of developing hereditary tumors based on their family background using IGV and RT-PCR.
Results: KRAS Q61K, which is found in many gastrointestinal cancers, was identified as a VUS by ClinVar, but this gene mutation was found to cause splicing. The cancer gene panel test of a 41-year-old male patient with paraganglioma identified succinate dehydrogenase complex, iron-sulfur subunit B (SDHB) G642T as a VUS. However, this mutation was later discovered to cause the splicing site to shift, preventing SDHB from translating from the correct mRNA. In addition, a cancer gene panel test of a 47-year-old patient with right breast cancer determined that breast cancer susceptibility gene 2 (BRCA2) 631 3A>T was a VUS. However, this mutation may create a splicing site, which means that the correct BRCA2 mRNA for BRCA2 is not produced.
Conclusions: The diagnosis of gene mutations based on the results of cancer gene panel testing may not always be correct, and a detailed examination of gene mutations is necessary. Our medical staff has performed cancer gene panel testing on approximately 5,500 cases of intractable malignant tumors to date and are investigating new treatments for these tumors. In this article, we discuss our experience with cancer gene panel testing as well as the problems encountered and new findings.
Keywords: Cancer gene panel; Pathogenic variant; VUS; Splicing
| Introduction | ▴Top |
Cancer treatment has involved the selection of antitumor drugs according to the cancer type in each organ. The effectiveness of antitumor drugs against various cancer types has been examined, and antitumor drugs have been prescribed according to the treatment guidelines for each cancer type [1]. However, the effectiveness of antitumor drugs, as defined by clinical practice guidelines, is not always known. Cancer cells are heterogeneous, and the unique characteristics of each individual cancer cell strongly affect the efficacy of the prescribed antitumor agent [2]. The “Precision Medicine Initiative” was announced in 2015 in the USA, which is developing into a new cancer treatment known as cancer genome medicine, which is not limited by the cancer type in each tissue [3, 4].
FoundationOne® CDx Cancer Genomic Profile (FMI, Cambridge, MA, USA) and OncoGuide™ NCC Oncopanel (Sysmex Corporation, Kobe, Hyogo, Japan) were approved in Japan, by the Ministry of Health, Labour and Welfare as cancer gene panel tests for solid tumors and have been covered by insurance in June 2019. Cancer gene panel testing has been initiated in Japan in clinical practice as a genomic cancer treatment. The genomic DNA extracted from sections of surgically removed tissues (solid tumors) are fixed in formalin and embedded in paraffin, then used for cancer gene panel testing. However, the genomic DNA in tissues fragments after long-term storage; therefore, tissue sections stored for more than 3 years cannot be used for cancer gene panel testing. Therefore, these cancer gene panel tests cannot be performed on patients who underwent surgical treatment more than 3 years ago, who were transferred from another hospital, or from whom tissue sections could not be obtained.
Many clinical studies have analyzed various parameters of the cell-free DNA in liquid biopsies (human blood samples), such as variations in bases, copy numbers, and structure; DNA fragmentation; and DNA methylation. Methods have been developed to detect and monitor the onset and progression of cancer based on various cell-free DNA parameters [5]. FoundationOne® CDx liquid (FMI) and Guardant360 CDx (Guardant Health, Inc., Palo Alto, CA, USA) have been approved by the Japanese Ministry of Health, Labour and Welfare as cancer gene panel tests for use with human blood samples. Nucleotide mutations and copy number changes were detected in the cell-free DNA obtained from human blood samples. Therefore, cancer gene panel testing using human blood samples is the method of choice when tissue sections removed by surgical treatment are not available or have been stored for more than 3 years.
Our medical staff performed cancer gene panel testing from 2019 to March 2025 to identify new treatment strategies for approximately 5,500 cases of progressive and metastatic malignant tumors. The gene mutations and increased copy numbers detected through cancer gene panel testing were compared with the data from databases such as ClinVar (National Center for Biotechnology Information) [6], OncoKB (an FDA-recognized human genetic variant database) [7], and Catalogue of Somatic Mutations in Cancer (COSMIC) [8] to determine their involvement in the onset or progression of malignant tumors. However, Kirsten rat sarcoma virus Protooncogene, GTPase (KRAS) gene mutations may be silent mutations or splicing donor sites, and such gene mutations do not affect the onset or progression of malignant tumors [9]. Therefore, our medical staff re-examined whether the gene mutations detected using cancer gene panel testing were silent mutations or splice donor sites.
Succinate dehydrogenase complex, iron-sulfur subunit B (SDHB) G642T was diagnosed as a variant of unknown significance (VUS) through cancer gene panel testing for a 41-year-old male patient with paraganglioma. However, this mutation was later discovered to cause a shift in the splicing site, preventing SDHB from being translated from the correct mRNA. In addition, breast cancer susceptibility gene 2 (BRCA2) 631 3A>T was identified as a VUS from the results of cancer gene panel testing of a 47-year-old patient with right breast cancer. However, this mutation created a splicing site that prevented the correct BRCA2 mRNA from being produced for BRCA2. As such, the medical information displayed in databases such as ClinVar is not always accurate; therefore, genome databases must also be used to examine the effects of gene mutations. Furthermore, the information in these databases may not be applicable to the onset or progression of malignant tumors in Asian populations as the data were obtained from Western patients. Therefore, medical information should be organized by race when creating a database.
| Materials and Methods | ▴Top |
Cancer gene panel profiling
This multicenter, retrospective, observational study included patients who received cancer genome treatment at various cancer medical facilities in Kyoto, Japan. This clinical study was performed in accordance with the principles of the Declaration of Helsinki and was approved by the Central Ethics Review Board of the National Hospital Organization Headquarters, Meguro, Tokyo, Japan (approval number: NHO R4-04; approval date: November 18, 2020) and the Kyoto University School of Medicine, Kyoto, Japan (approval number: M237; approval date: August 24, 2022). Written informed consent was obtained from all participants.
Cancer genome medicine was applied using cancer gene panel testing, which was approved by the Japanese Ministry of Health, Labor, and Welfare on June 3, 2019. Cancer gene panel testing involved the use of the OncoGuide™ NCC Oncopanel gene mutation analysis set (Sysmex Corporation, Kobe, Hyogo, Japan) and FoundationOne CDx cancer genome test (Foundation Medicine, Inc., Cambridge, MA, USA).
The mutation information of 324 genes was analyzed using the FoundationOne CDx cancer genome profile, using the genomic DNA of the tumor tissue samples (including cytology samples) obtained from the patients with solid cancer and in the cell-free DNA of the plasma separated from the whole-blood samples of the patients with solid cancer. The mutation information of 114 genes was analyzed using the OncoGuide™ NCC Oncopanel with the genomic DNA of the tumor tissue specimens (including cytology specimens) obtained from the patients with solid cancers. In addition, the program was used to analyze the mutation information of 114 genes in the cell-free DNA of the plasma separated from the whole-blood samples of patients with solid cancer. We determined whether the genomic mutations in the tumor tissue samples were germline mutations.
A total of 5,504 novel treatments were investigated using cancer genome panel testing (FoundationOne® CDx test: n = 4,135; OncoGuide™ NCC Oncopanel test, Riken Genesis, Yokohama, Kanagawa, Japan, n = 1,369) at Japanese national universities from December 2019 to May 2025.
Analysis of the three-dimensional structure of the binding site between SDHB and SDHAF2
Spanner is a structural homology modeling pipeline that threads a query amino-acid sequence onto a template protein structure using protein structure Protein Data Bank (PDB) ID 3SFE as template protein structure. Spanner is unique in that it handles gaps by spanning the region of interest using fragments of known structures. To create a model, you must provide a template structure, as well as an alignment of the query sequence you wish to model onto the template sequence. Spanner will replace mismatched residues, and fill any gaps caused by insertions or deletions (Supplementary Material 1, wjon.elmerpub.com).
Consent for germline testing involving sensitive genetic information
When the results of the cancer genome panel test indicated the possibility of hereditary cancer (familial cancer), our medical staff confirmed with the patient whether they wanted the test results to be communicated to them or to their family. In addition, our medical staff informed the patient that if hereditary cancer was diagnosed, her son would need to undergo cancer surveillance. In addition, our medical staff informed patients of the need for their children to undergo cancer surveillance when hereditary cancer was diagnosed. Furthermore, we, the medical staff, changed the number so that the patient could not be identified, and confirmed the patient’s consent to reporting the results of the cancer genome panel test as medical information at academic conferences or in medical journals.
| Results | ▴Top |
The results obtained from the cancer gene panel tests performed by our medical staff are mainly based on the medical information in ClinVar. However, some of the medical information in ClinVar does not reflect specific pathogenic variants in Asian, including Japanese, populations [10]. Therefore, the detection of genetic cancers in a family may be missed. Therefore, medical staff must reverify the medical information in ClinVar.
Case 1
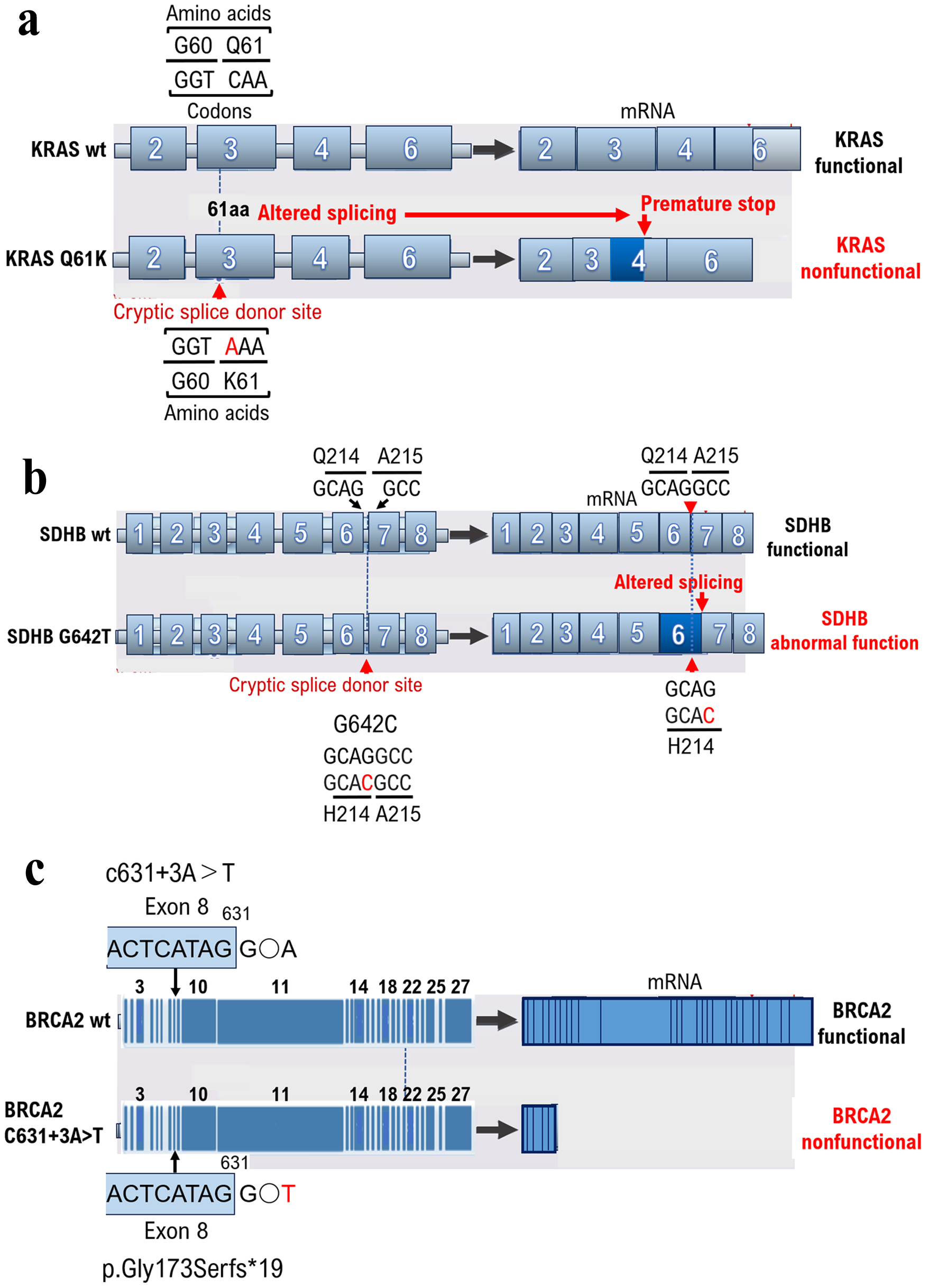
A silent mutation is a base mutation that changes the DNA sequence of a gene but does not result in a change in the amino acid sequence of a protein [11] and has no effect on the organism. Missense mutations alter a single amino acid and have serious consequences if the change alters the function of the protein. However, the KRAS mutations detected through cancer gene panel testing at cancer genome medical facilities may be classified as VUSs using ClinVar. However, even silent KRAS gene mutations create a base sequence that indicates the start of a new intron (Intron base sequence: Intron locations must be determined to remove introns via RNA splicing. Rules exist for the base sequence of introns, and almost all introns (> 99%) start with GU and end with AG, which is called the GU-AG rule. The 5' side of the GU sequence (the 5' end of the intron) is called the 5' splice site, and the 3' side of the AG sequence (the 3' end of the intron) is called the 3' splice site. Another important sequence is the branch site, located 18 - 40 nucleotides upstream of the 3’ splice site. The consensus sequence for the branch site in yeast is UACUAAC and YNYYRAY in animals, with adenine (A), shown in red, being the branch site. Downstream of the branching site is a region of consecutive pyrimidines (C and U).), which can lead to splicing (Fig. 1a, Supplementary Materials 2 and 3, wjon.elmerpub.com). The primary protein structure is substantially altered in such cases; therefore, the gene mutation is not a VUS but a pathogenic variant. Therefore, caution is required when determining VUSs using ClinVar, as gene mutations resulting from gene mutation mechanisms may become splicing sites.
 Click for large image |
Figure 1. Base mutations can result in intron start signals, resulting in unexpected splicing. The results of cancer genome panel testing depend on a database built from the medical information of patients in Europe and the United States. Human genes differ depending on race; therefore, test results are not always correct. Unexpected splicing can occur because of base mutations that can cause intron start signals. (a) A silent KRAS gene mutation creates a base sequence that indicates the start of a new intron, which can lead to splicing. (b) The SDHB gene mutation G642C was determined to be a VUS from the results of a cancer gene panel test for a 41-year-old male patient with PPGL. The patient had no known family history of PPGL; however, PPGL onset occurred early in life. As a result, the mutation of the base G642C in the gene could have caused splicing abnormalities although the amino acid Q214H mutation was a VUS, as determined when checking the annotation content of the results of the cancer gene panel test. (c) Multiple family members of this patient had breast cancer, suggesting hereditary breast and ovarian cancer. Genetic testing for breast cancer susceptibility genes (BRCAI and BRCA2) was performed before the onset of ovarian cancer using BRAC Analysis (Myliad Co., Ltd., MA, USA). The c631+3A>T mutation was identified as a VUS based on the results of ClinVar analysis. However, we found that the c631+3A>T base mutation resulted in p.Gly173SerFs*19, which was diagnosed as a pathogenic variant when examined using the Medical Genomics Reviews Knowledge base for genomic medicine in Japanese (MGenReviews; National Institute of Global Health and Medicine, National Health Research Organization/National Center for Global Health and Medicine), a database that compiles the results of analyses of mutations specific to Japanese people. |
Based on the results of cancer gene panel testing, KRAS K61 mutation was reported as a VUS, and therefore anti-epidermal growth factor receptor (EGFR) antibodies or anti-EGFR drugs were recommended. After further review, it was determined that KRAS K61 mutant is an inactive form of KRAS, and therefore anti-rapidly accelerated fibrosarcoma (RAF) drugs or anti-serine/threonine kinase drugs were recommended.
Case 2
Pheochromocytoma and paraganglioma (PPGL) are rare endocrine hypertensive diseases that are often curable [12]. However, the signs and symptoms of PPGL are nonspecific and diverse. Furthermore, PPGL are difficult to differentiate from panic disorders, cardiovascular diseases, and other diseases. Therefore, medical staff should consider PPGL during diagnosis. PPGL were incidentally discovered during imaging tests for other diseases. Test results must be carefully examined using random urine metanephrine and normetanephrine measurements, magnetic resonance imaging, and metaiodobenzylguanidine scintigraphy because PPGL are asymptomatic [13]. Genetic mutations are present in approximately 30% of PPGL cases. Mutation testing should be performed for the multiple endocrine neoplasia type 2A gene for suspected von Hippel-Lindau disease, particularly in cases where tumors occur in families with PPGL, tumors develop at a young age, and hereditary tumor syndrome is suspected [13, 14].
Testing for SDHB mutations is recommended for tumors with no multiple endocrine neoplasia type 2A gene mutations and von Hippel-Lindau disease is ruled out. SDHB is a subunit of succinate dehydrogenase involved in the TCA cycle [15]. The SDHB mutation G642C was determined to be a VUS as a result of a cancer gene panel test (GenMinTOP) on a 41-year-old male patient with PPGL (Fig. 1b). PPGL onset was early in life, despite the lack of known family history of PPGL; therefore, we performed further genomic analyses. As a result, we found that mutation of the base G642C in the gene could have caused splicing abnormalities, when checking the annotation content of the results of the cancer gene panel test, although the amino acid Q214H mutation was a VUS (Fig. 1b, Supplementary Materials 3 and 4, wjon.elmerpub.com). The results of RNA sequencing were analyzed using Integrative Genomics Viewer (IGV, a tool for visualizing next-generation sequencing mapping results and annotation information. IGV has been used for confirming the level of gene expression from RNA-seq mapping results, searching for single-nucleotide polymorphisms from whole-genome sequencing results, and narrowing candidate regions for cis-regulatory sequences from ChiP-seq and ATAC results.), and splicing was confirmed to have occurred due to base mutations. As in this case, splicing may occur because of base mutations even if the amino acid mutation (Q214H) is determined to be a VUS using ClinVar. Therefore, the presence of pathogenic variants should be reconfirmed based on medical information such as the age at the onset of malignant tumors and family history. To examine the extent to which the binding ability between SHDB and SDAF1 is affected by the glutamine-to-histidine mutation, we performed in silico analysis. To examine the extent to which the binding ability between SHDB and SDAF1 is affected by the glutamine-to-histidine mutation, we performed in silico analysis. In silico analysis showed that glutamine 214 of SHDB has affinity for histidine 156 of SDAF2. The affinity between glutamine 214 of SHDB and tyrosine 156 of SDAF2 was found to be -17.93 kcal/mol. On the other hand, although the histidine 214 of the SHDB mutant has affinity for the tyrosine 156 of SDAF2, the affinity between histidine 214 of SHDB and tyrosine 156 of SDAF2 was found to be weak, at -6.31 kcal/mol. In other words, the results of in silico analysis revealed that the SHDB 214His mutant may have a much weaker binding affinity to SDAF2 than the wild-type SHDB.
Based on the results of cancer gene panel testing, the SDHB H214 mutation was reported as a VUS, and therefore the pheochromocytoma/paraganglioma in this case was not determined to be hereditary pheochromocytoma/paraganglioma syndrome (HPPS). After further investigation, it was found that the binding affinity of the SDHB H214 mutant to SDFA2 was weak, and therefore the biological activity of the SDHB H214 mutant was lost. Therefore, families with SDHB H214 variants are considered to have HPPS. Therefore, it was suggested that the patient’s family, primarily his son/daughter, undergo genetic testing for the SDHB H214 mutation and be monitored for the onset of HPPS.
Case 3
Approximately 5-10% of breast cancers are hereditary [16]; however, a detailed professional evaluation is required to verify if the breast cancer case is hereditary. Hereditary breast cancer is possible in people with early-onset breast cancer (onset age of 35 years or younger), bilateral or multiple breast cancer, male breast cancer, and both ovarian and breast cancer, even in the absence of a family history of breast or ovarian cancer [17]. In our case, multiple family members had breast cancer, suggesting hereditary breast and ovarian cancer. Genetic testing for breast cancer susceptibility genes (BRCA1 and BRCA2) was performed before the onset of ovarian cancer (HBOC) using BRAC Analysis (Myliad Co., Ltd., MA, USA) to prescribe risk-reducing salpingo-oophorectomy [18] and a PARP inhibitor (i.e., olaparib). The c631+3A>T mutation was identified as a VUS through ClinVar analysis (Fig. 1c, Supplementary Materials 3 and 5, wjon.elmerpub.com). However, we found that the c631+3A>T base mutation resulted in p.Gly173SerFs*19, which was identified as a pathogenic variant when examined using the Medical Genomics Reviews Knowledge base for genomic medicine in Japanese (MGenReviews; National Institute of Global Health and Medicine, National Health Research Organization/National Center for Global Health and Medicine) [19], a database that compiles the results of analyses of mutations specific to Japanese people.
Based on the results of cancer gene panel testing, the BRCA2 C631+3A>T mutation was reported as a VUS, and therefore platinum agents or poly (ADP-ribose) polymerase (PARP) inhibitors were not recommended. After further investigation, it was determined that the BRCA2 C631+3A>T variant results in a deletion of exon 9 and beyond. Therefore, families of patients with the BRCA2 C631+3A>T mutation were deemed to have hereditary breast and ovarian cancer (HBOC). In addition, it was suggested that the patient’s family, mainly his son/daughter, undergo genetic testing for the BRCA2 C631+3A>T mutation and be monitored for the development of HBOC.
| Discussion | ▴Top |
FoundationOne® CDx Cancer Genomic Profile (FMI, Cambridge, MA, USA) and OncoGuide™ NCC Oncopanel (Sysmex Corporation, Kobe, Hyogo, Japan) were approved by the Ministry of Health, Labour and Welfare in Japan as cancer gene panel tests for solid tumors and were covered by insurance as of June 2019. Subsequently, cancer gene panel testing was initiated in Japan to guide genomic cancer treatment. Our medical staff performed cancer gene panel testing from 2019 to March 2015 to identify new treatment strategies for approximately 5,500 cases of progressive and metastatic malignant tumors. A silent mutation does not result in a change in the amino acid sequence of a protein, even if occurring in the DNA sequence of a gene, and does not affect the organisms. Missense mutations alter a single amino acid and may have serious consequences if the change alters protein function. In addition, the gene mutations detected by cancer gene panel testing at cancer genome medical facilities have been determined as VUSs using ClinVar. However, new splicing may occur due to genetic mutation even if a mutation is silent due to genetic mutation. The primary structure of the protein considerably changes in such cases; these gene mutations are pathogenic variants rather than VUSs. Therefore, genetic mutations are resulting in new splice sites.
An integrated approach that includes both epigenetic and genetic factors has been developed for identifying early cancer signals [20]. Another approach that combines multiple cell-free DNA features and provides details on cancer onset and genomes was devised to improve the detection of cancer onset [21, 22]. Therefore, the cancer genome, in addition to surgically removed tissues, is tested using liquid biopsies in cancer genome medicine. A Korean research team is investigating a next-generation sequencing platform based on cell-free DNA analysis, the AlphaLiquid® Screening Platform [23]. Whole-genome methylation sequencing (WGMS) was used to quantify the DNA methylation patterns in the CpG regions throughout the genome. The results of the clustering analysis of data obtained from patients with cancer and healthy individuals enabled their differentiation as well as the estimation of the tissues in which the cancer originated. This platform simultaneously analyzes whole-genome DNA methylation sites, copy number alterations, and fragment patterns in noninvasively isolated cell-free DNA samples using WGMS. In particular, this platform employs enzymatic methylation conversion technology for DNA methylation analysis, which minimizes the damage caused by fragmentation and coverage bias, and enables the acquisition of highly accurate data [24].
Currently, cancer gene panel testing is used to detect pathogenic variants associated with tumor development in samples of tumor tissue removed by surgical treatment or blood collected from patients. Furthermore, antitumor compounds that specifically recognize and bind to these pathogenic variants are being explored. In many cases, when tumor-specific gene mutations are found in tumor tissue removed by surgical treatment or in blood samples taken from the patient using cancer gene panel testing, these mutations are examined using databases developed in Europe and the United States, such as ClinVar, COSMIC, OncoKB, and VRsome, to determine whether they are pathogenic variants. Because the genes of Westerners differ from those of Asians, including Japanese, genetic background can raise questions about whether the gene mutations detected by cancer gene panel testing are VUS. In such cases, at the request of medical staff, the testing company may re-examine the meaning of the genetic mutation, and the genetic mutation may be recognized as a pathogenic variant specific to Japanese people. If a genetic mutation identified as a VUS by ClinVar is later determined to be a pathogenic variant by detailed examination by the testing company, this means that new antitumor drug options are available to patients.
Gene mutations play a major role in the development of cancer and malignant tumors. However, the base sequences of genes change within the human body to adapt to long-term differences in the environment, such as differences in diet and temperature [25]. Consequently, the gene sequences differ among races, suggesting the involvement of pathogenic variants during the development and progression of cancer and malignant tumors differs among races. The databases used in clinical practice to re-examine pathogenic variants include ClinVar, OncoKB, and COSMIC, which are based on data obtained from Western patients. Therefore, a database of the pathogenic variants for each race must be built. To achieve this, medical staff, together with protein engineering scientists, must construct the three-dimensional structure of protein mutants through in silico analysis and examine their physiological activity. New treatments for progressive and metastatic malignant tumors are being developed using cancer gene panel testing. However, the types of antitumor agents that can be used against pathogenic variants, which are identified based on various factors, are limited. Several clinical studies have investigated the efficacy of new regimens for advanced and metastatic malignant tumors to improve current cancer treatment.
Conclusion
Cancer gene panel testing has led to considerable advances in personalized cancer treatment. However, the results of cancer genome panel testing depend on a database built from the medical information of patients in Europe and the United States. Human genes differ with race; therefore, test results are not always correct. Unexpected splicing can occur because of base mutations that can cause intron start signals. Therefore, medical staff must carefully examine the test results. Additionally, database of cancer genomes that includes gene mutations specific to Asian populations must be built.
| Supplementary Material | ▴Top |
Suppl 1. Analysis of the three-dimensional structure of the binding site between SDHB and SDHAF2.
Suppl 2. The structure and functional sites of KRAS.
Suppl 3. Comparison of wild-type and mutant sequences.
Suppl 4. The SDHB G642T variant is unable to bind to the SDAF2 due to the mutation. Therefore, this mutant cannot perform its normal function.
Suppl 5. Diagram of the positions of identified domains in human BRCA2 and of the 27 exons that encode human BRCA2 product.
Acknowledgments
The authors want to thank Dr. Yoshiihiro Kawaoka at The Institution of Medical Science, The University of Tokyo for providing clinical research information. The authors also want to acknowledge all medical staff for clinical research at Kyoto University School of Medicine and the National Hospital Organization Kyoto Medical Center. We also appreciate Chugai Pharma Manufacturing Co., Ltd. (Kitaku, Tokyo, Japan) and Sysmex Corporation (Kobe, Hyogo, Japan) for providing medical imformation.
Financial Disclosure
This clinical research was performed using research funding from the following: Japan Society for Promoting Science for TH (grant no. 19K09840), START-program Japan Science and Technology Agency (JST) for TH (grant no. STSC20001), National Hospital Organization Multicenter Clinical Study for TH (grant no. 2019-Cancer in general-02), and Japan Agency for Medical Research and Development (AMED) (grant no. 22ym0126802j0001), Tokyo, Japan. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
None to declare.
Informed Consent
Not applicable.
Author Contributions
TH and IK were involved in the study design, data collection, data review and interpretation, literature search, and manuscript writing.
Data Availability
Data are available on various websites and have also been made publicly available. More information can be found in the first paragraph of the Results section. The transparency document associated with this article can be found in the online version at https://center6.umin.ac.jp/cgi-open-bin/ctr/ctr_view.cgi?recptno=R000044182.
| References | ▴Top |
- Califf RM, Zarin DA, Kramer JM, Sherman RE, Aberle LH, Tasneem A.
Characteristics of clinical trials registered in ClinicalTrials.gov, 2007-2010. JAMA.
2012;307(17):1838-1847.
doi pubmed - Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer.
Nature. 2019;575(7782):299-309.
doi pubmed - Modell SM, Citrin T, Kardia SLR. Laying anchor: inserting precision
health into a public health genetics policy course. Healthcare (Basel). 2018;6(3):93.
doi pubmed - Kurian AW, Abrahamse P, Furgal A, Ward KC, Hamilton AS, Hodan R,
Tocco R, et al. Germline genetic testing after cancer diagnosis. JAMA.
2023;330(1):43-51.
doi pubmed - Cisneros-Villanueva M, Hidalgo-Perez L, Rios-Romero M, Cedro-Tanda A,
Ruiz-Villavicencio CA, Page K, Hastings R, et al. Cell-free DNA analysis in current cancer
clinical trials: a review. Br J Cancer. 2022;126(3):391-400.
doi pubmed - https://www.ncbi.nlm.nih.gov/clinvar/?term=.
- https://www.oncokb.org/.
- https://cancer.sanger.ac.uk/cosmic/login.
- Lu J, Zhou C, Pan F, Liu H, Jiang H, Zhong H, Han B. Role of silent
mutations in KRAS -mutant tumors. Chin Med J (Engl). 2025;138(3):278-288.
doi pubmed - Jiang Y, Li R, Li X. [Report of a child with Bainbridge-Ropers
syndrome due to a novel variant of ASXL3 gene and a literature review]. Zhonghua Yi Xue Yi Chuan
Xue Za Zhi. 2024;41(8):966-972.
doi pubmed - Kobayashi Y, Chhoeu C, Li J, Price KS, Kiedrowski LA, Hutchins JL,
Hardin AI, et al. Silent mutations reveal therapeutic vulnerability in RAS Q61 cancers. Nature.
2022;603(7900):335-342.
doi pubmed - Chung SM. Screening and treatment of endocrine hypertension focusing
on adrenal gland disorders: a narrative review. J Yeungnam Med Sci.
2024;41(4):269-278.
doi pubmed - Que FVF, Ishak NDB, Li ST, Yuen J, Shaw T, Goh HX, Zhang Z, et al.
Utility of whole-body magnetic resonance imaging surveillance in children and adults with cancer
predisposition syndromes: a retrospective study. JCO Precis Oncol. 2025;9:e2400642.
doi pubmed - Shirali AS, Clemente-Gutierrez U, Huang BL, Lui MS, Chiang YJ,
Jimenez C, Fisher SB, et al. Pheochromocytoma recurrence in hereditary disease: does a
cortical-sparing technique increase recurrence rate? Surgery. 2023;173(1):26-34.
doi pubmed - Zhao X, Sun K, Lan Z, Song W, Cheng L, Chi W, Chen J, et al.
Tenofovir and adefovir down-regulate mitochondrial chaperone TRAP1 and succinate dehydrogenase
subunit B to metabolically reprogram glucose metabolism and induce nephrotoxicity. Sci Rep.
2017;7:46344.
doi pubmed - Lux MP, Fasching PA, Beckmann MW. Hereditary breast and ovarian
cancer: review and future perspectives. J Mol Med (Berl). 2006;84(1):16-28.
doi pubmed - Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M,
Timmerman MM, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2
among Ashkenazi Jews. N Engl J Med. 1997;336(20):1401-1408.
doi pubmed - Lee EG, Kang HJ, Lim MC, Park B, Park SJ, Jung SY, Lee S, et al.
Different patterns of risk reducing decisions in affected or unaffected BRCA pathogenic variant
carriers. Cancer Res Treat. 2019;51(1):280-288.
doi pubmed - https://mgen.ncgm.go.jp/.
- Hayashi T, Konishi I. Correlation of anti-tumour drug resistance with
epigenetic regulation. Br J Cancer. 2021;124(4):681-682.
doi pubmed - Siejka-Zielinska P, Cheng J, Jackson F, Liu Y, Soonawalla Z, Reddy S,
Silva M, et al. Cell-free DNA TAPS provides multimodal information for early cancer detection.
Sci Adv. 2021;7(36):eabh0534.
doi pubmed - Jamshidi A, Liu MC, Klein EA, Venn O, Hubbell E, Beausang JF, Gross
S, et al. Evaluation of cell-free DNA approaches for multi-cancer early detection. Cancer Cell.
2022;40(12):1537-1549.e1512.
doi pubmed - https://www.imbdx.com/eng/about/profiling.php.
- Vaisvila R, Ponnaluri VKC, Sun Z, Langhorst BW, Saleh L, Guan S, Dai
N, et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from
picograms of DNA. Genome Res. 2021;31(7):1280-1289.
doi pubmed - Ames BN. Dietary carcinogens and anti-carcinogens. J Toxicol
Clin Toxicol. 1984;22(3):291-301.
doi pubmed
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
World
Journal of Oncology is published by Elmer Press Inc.