| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://wjon.elmerpub.com |
Original Article
Volume 16, Number 5, October 2025, pages 457-470

Liquid Plasma Induces Necroptosis Without Inflammatory Responses in Head and Neck Cancer Cells
Jae Hoon Choia, b, d, Sungryeal Kimc, d, Yun Sang Leea, d, Young Suk Youa, Jeon Yeob Janga, Yoo Seob Shina, Chul-Ho Kima, b, e
aDepartment of Otolaryngology, School of Medicine, Ajou University, Suwon 16499, Korea
bDepartment of Molecular Science and Technology, Ajou University, Suwon 16499, Korea
cDepartment of Otolaryngology, College of Medicine, Inha University, Incheon 22332, Korea
dThese authors contributed equally to this work.
eCorresponding Author: Chul-Ho Kim, Department of Otolaryngology, School of Medicine, Ajou University, Yeongtong-gu, Suwon 16499, Korea
Manuscript submitted March 18, 2025, accepted September 1, 2025, published online September 17, 2025
Short title: Plasma Induces Necroptosis Without Inflammation
doi: https://doi.org/10.14740/wjon2579
| Abstract | ▴Top |
Background: Several types of regulated cell deaths are known, including apoptosis, necroptosis, autophagy, ferroptosis, and pyroptosis. Among these types of cell deaths, apoptosis is induced by many cancer therapeutic agents. In the case of resistance, however, induction of other regulated cell death, such as necroptosis, are required. Liquid plasma, which is prepared by treatment of non-thermal plasma to solution, induces various types of regulated cell death via reactive oxygen and nitrogen species.
Methods: Liquid plasma was generated by N2/Ar plasma treatment in culture medium (minimum essential medium (MEM), Dulbecco’s modified Eagle medium (DMEM), or Roswell Park Memorial Institute (RPMI)-1640) for 120 s per milliliter of medium (2 cm). Cell viability was determined using Cell Counting Kit-8 (CCK8), and apoptosis was determined by terminal deoxynucleotidyl transferase deoxyuridine triphosphate (dUTP) nick end labeling (TUNEL) assay. Tumor necrosis factor alpha (TNF-α), cycloheximide (CHX), and zVAD-fmk were used to induce necroptosis in head and neck squamous cell carcinoma (HNSCC) cells, and necroptosis inhibitors, such as necrostatin-1 (Nec-1, 50 µM), GSK872 (10 µM), and necrosulfonamide (NSA, 2 µM) were used to inhibit necroptosis. Statistical comparisons between groups were carried out using the Student’s t-test.
Results: Here, we determined the type of cell death induced by liquid plasma in head and neck cancer (HNC) cells. Our results show that liquid plasma caused necroptosis in HNC cells, and peroxynitrite in the liquid plasma might be involved in the cell death. The expression of inflammation-related molecules, including nuclear factor kappa B (NF-κB), interleukin (IL)-6, and mitochondrial antiviral signaling proteins, were detected in HNC cells, and treatment of HNC cells with liquid plasma decreased their expression.
Conclusions: These results suggest that liquid plasma could be used to treat HNC by inducing necroptosis without inflammatory responses. In this study, we demonstrated that liquid plasma treatment may kill HNC cells without causing necroptosis-induced inflammation and inflammation-mediated diseases.
Keywords: Liquid plasma; Necroptosis; Head and neck cancer; Inflammation; Treatment
| Introduction | ▴Top |
Since Lockshin et al mentioned programmed cell death in 1964 [1], various types of programmed cell death have been discovered, including apoptosis and necroptosis [2]. Unlike apoptosis, caspase-dependent cell death, necroptosis, also called programmed necrosis, induces inflammatory immunogenic cell death through receptor-interacting protein kinase 1 (RIP1)-RIP3-mixed lineage kinase domain-like protein (MLKL) instead of caspases [3]. Briefly, necroptosis in cancer cells requires RIP3, and its kinase activity is required to form a stable necrosome complex that induces necroptosis [4, 5]. Upon activation by tumor necrosis factor alpha (TNF-α), RIP1 binds to and activates RIP3, which in turn activates MLKL via phosphorylation, leading to the formation of a necrosome complex. Necroptosis involves translocation of MLKL to the plasma membrane [6]. This molecular mechanism of necroptosis has recently been considered a promising strategy for cancer treatment. Some cancer chemotherapy drugs cause apoptosis [7]; however, if mutations in proteins involved in the apoptotic pathway or increased expression of antiapoptotic proteins occur, the chemotherapy will fail. Previous studies have shown increased production of Bcl-2, Bcl-xL, or FLIP, which are anti-apoptotic proteins, or mutations in p53, Bax, Fas, or caspases in cancer cells [8, 9]. Therefore, other chemotherapeutic drugs that cause different forms of cell death may need to be developed. Indeed, in several carcinomas, including breast, leukemia, ovarian, and colorectal cancers, resistant cancer cells can be killed through necroptosis during apoptotic therapy [9-12].
Necroptosis is a programmed form of cell death that is caspase-independent and morphologically similar to necrosis. Necrosis may occur in cells where apoptosis cannot be induced. Therefore, necroptosis could act as an alternative method of programmed cell death to overcome resistance to apoptosis. Unlike apoptosis, necroptosis induces inflammatory responses by releasing damage-associated molecular pattern proteins. Therefore, necroptosis is involved in many inflammatory diseases, such as Crohn’s disease and pancreatitis [13, 14], and inhibition of necroptosis-induced inflammatory responses might be critical in preventing or treating these diseases. Therefore, the induction of necroptosis without inflammatory responses may be a promising strategy to treat cancer.
Plasma is the fourth state of matter along with solid, liquid, and gas, and it is created by applying energy to gas. Reactive oxygen and nitrogen species (RONS), radiation, and electronic fields generated by non-thermal atmospheric plasma are useful in medical fields for various applications, such as disinfection, dental care, skin disease, chronic wounds, cosmetics, tissue regeneration, and cancer therapy [13]. RONS not only stimulate cell proliferation but also induce cell death if intracellular RONS levels reach a toxic threshold. Because the intracellular levels of RONS in cancer cells are already high, induction of RONS would not affect normal cells but would kill cancer cells [14]. This selectivity of non-thermal plasma (NTP) has attracted interest, and its anticancer effect has been reported in several carcinomas, including brain, skin, breast, head and neck, blood, ovary, gastrointestinal, bone, and lung [15-25].
NTP treatment has been shown to induce multiple regulated cell death (RCD) pathways [26], including apoptosis [25, 27, 28] autophagic cell death [29], pyroptosis [30], ferroptosis [31], and necroptosis [32], depending on the cancer cell type and molecular context, and the gas composition used for plasma generation [33]. Notably, apoptosis and pyroptosis share caspase-3 activation as a common feature; however, the transition to pyroptosis occurs when cancer cells overexpress gasdermin E (GSDME), which forms membrane pores to induce pyroptotic cell death [30]. Caspase-8 serves as a key regulator of apoptosis and necroptosis, activated through death receptor signaling to trigger apoptosis by cleaving downstream effectors like caspase-3 [34]. However, FLICE-like inhibitory protein (cFLIP), often overexpressed in cancer cells, inhibits procaspase-8 activation by forming a heterodimer, which interacts with RIPK1 and RIPK3 to drive necrosome assembly and necroptosis [35]. Similarly, ferroptosis is more likely in cancer cells with high levels of polyunsaturated fatty acids and low expression of the cystine transporter solute carrier family 7 member 11 (SLC7A11 also called xCT) [31], while necroptosis is associated with the p-MLKL activation and elevated MLKL expression levels [36]. These observations underscore that NTP-induced cancer cell death is influenced not only by plasma conditions but also by the molecular profile of the target cancer cells.
In this study, we investigated whether liquid plasma (LP), which is generated by treatment of NTP in solution, could trigger necroptosis in head and neck cancer (HNC) cells. Our results demonstrate that LP treatment induced necroptosis. In addition, it was discovered that LP-induced necroptosis does not promote inflammation through degradation of mitochondrial antiviral signaling (MAVS) protein and inhibition of nuclear factor kappa B (NF-κB) activation. These results suggest that LP treatment could be a new option for HNC therapy without inflammation, which is involved in many inflammatory diseases, such as Crohn’s disease, pancreatitis, and neurodegenerative disorders.
| Materials and Methods | ▴Top |
Cells culture and reagents
HNC cell lines, including FaDu cells (human hypopharyngeal cancer, American Type Culture Collection (ATCC), HTB-43) and SNU1041 cells (human hypopharyngeal cancer, Korean Cell Line Bank (KCLB), 01041.1), were obtained from their respective sources. Human dermal fibroblasts (HDFa, PCS-201-012) were purchased from the ATCC (Manassas, VA, USA). MSKQLL1 cells (human oral cavity cancer) [37] and SCCQLL1 cells (a squamous cell carcinoma cell line originating from metastatic lymph nodes in oral cancer) [38] were generously provided by Prof. Se-Heon Kim (Yonsei University, Korea). 293T cells (ATCC, CRL-3216) were supplied by Prof. Hyeseong Cho (Ajou University, Korea). HN3 cells (human laryngeal cancer) and HN6 cells (human floor-of-mouth cancer) [39] were obtained from Prof. Sang Yoon Kim (Asan Medical Center, Korea).
FaDu, HN3, HN6, and SCCQLL1 cells were cultured in minimum essential medium (MEM) (Welgene, Daegu, Korea) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA), 1% antibiotic-antimycotic containing penicillin, streptomycin, and amphotericin B (Gibco, 15240062), and 1% sodium pyruvate (Gibco, 11360-070). SNU1041 cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 (Welgene, Daegu, Korea) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA), and 1% antibiotic-antimycotic (Gibco, 15240062). MSKQLL1 cells were cultured in Dulbecco’s modified Eagle medium (DMEM)/F12 (Welgene, Daegu, Korea) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA) and 1% antibiotic-antimycotic (Gibco, 15240062). 293T cells and fibroblasts were cultured in DMEM (Welgene, Daegu, Korea) supplemented with 10% fetal bovine serum (Gibco, Carlsbad, CA, USA), 1% antibiotic-antimycotic (Gibco, 15240062). Cells were incubated at 37 °C in 5% CO2 under humidified conditions.
Recombinant human TNF-α was purchased from PeproTech (Rockville, MD, USA). The pan-caspase inhibitor zVAD-fmk, RIP1 inhibitor necrostatin-1 (Nec-1), RIP3 inhibitor (GSK872), and MLKL inhibitor (NSA) were purchased from Selleckchem (Houston, TX, USA). N-acetylcysteine (NAC, A9165), manganese (III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP, 475870), sodium nitrate (7631-99-4), and sodium nitrite (7632-00-0) were purchased from Sigma-Aldrich. Cycloheximide (CHX, 2112) was purchased from Cell Signaling Technology (USA). Antibodies against RIP1 (Cell Signaling Technology, #3493), p-RIP1 Ser161 (Thermo Fisher Scientific, PA5-105640), RIP3 (Cell Signaling Technology, 13526), p-RIP3 Ser227 (Cell Signaling Technology 93654), MLKL (Cell Signaling Technology, 91689), cleaved caspase 3 (Cell Signaling Technology, 9661), cleaved caspase 8 (Cell Signaling Technology, 9496), cleaved caspase 9 (Cell Signaling Technology, 9505), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Cell Signaling Technology, 2118), MAVS (Cell Signaling Technology, 3993), NF-κB (Cell Signaling Technology, 8242), and p-NF-κB Ser536 (Cell Signaling Technology, 3033) were purchased from Cell Signaling Technology (USA). Anti-phospho-MLKL Ser358 (Abcam, ab187091) was purchased from Abcam (Cambridge, UK).
N2/Ar plasma characteristics and preparation of LP
In this study, as reported in previous experiments [40], a spray-type NTP system was developed and manufactured by adding Ar to N2 plasma using an N2/Ar plasma machine. The N2 gas of the machine was maintained at a flow rate of 200 standard cubic centimeters per minute (sccm), and the Ar gas was maintained at a flow rate of 1,500 sccm. We treated N2/Ar plasma in culture medium (MEM, DMEM, or RPMI-1640) according to the manufacturer’s protocol for 120 s per milliliter of medium (2 cm). After the culture medium was removed, cells were treated with 100 µL for 96-well plates, 1 mL for 12-well plates, and 3 mL for 60-mm dishes.
Cell counting
Cells were treated with LP for 0, 24, and 36 h and subsequently counted. Each cell line was washed with phosphate-buffered saline (PBS), detached using 0.25% trypsin- ethylenediaminetetraacetic acid (EDTA) (Gibco, 25200-056), and centrifuged at 1,300 rpm for 3 min. Trypsin solution was removed, and the cells were resuspended in 1 mL of PBS. Cell suspension (10 µL) was mixed 1:1 with 0.4% trypan blue (Gibco, 15250-061) and counted using a hemocytometer.
Cell viability assay
HNC cells were seeded in 96-well plates at a density of 5 × 103 cells/well and incubated for 12 h. Following incubation, the culture medium was replaced with plasma-treated medium, and the cells were further incubated for 16 - 24 h. Cell viability was measured by using the Cell Counting Kit (CCK-8; Dojindo, Tokyo, Japan) according to the manufacturer’s instructions. The CCK-8 solution (10 µL per well) was added, and the plate was incubated for 1 h. We measured cell viability at 450 nm, as specified in the CCK-8 assay protocol, with 600 nm as the reference wavelength to reduce background noise, using an Epoch Microplate Spectrophotometer (BioTek Instruments, Winooski, VT, USA). Each experiment was repeated in triplicate.
To assess the selective effect of LP on cancer cells compared to normal cells, FaDu, HN3, and SNU1041 (HNC cell lines) as well as fibroblasts and HaCaT cells were exposed to LP for 16 - 24 h, and their viability was evaluated using the CCK-8 assay.
To determine the role of necroptosis inhibitors, FaDu cells were pretreated with Nec-1 (50 µM), GSK872 (10 µM), or NSA (2 µM) for 1 h prior to LP treatment. After incubation for 16 - 24 h, cell viability was measured.
To investigate the role of peroxynitrite in LP-induced necroptosis, FaDu cells were pretreated with the peroxynitrite scavenger MnTBAP or NAC for 1 h before exposure to LP. Cell viability was assessed 16 - 24 h post-treatment.
Three-dimensional (3D) spheroid culture
HNC cells were added to a round 96-well plate with 1 × 104 cells/well. Following centrifugation at 3,000 rpm for 15 min at 37 °C, the cells were incubated for 7 days at 37 °C and 5% CO2 under humidified conditions.
Terminal deoxynucleotidyl transferase deoxyuridine triphosphate (dUTP) nick end labeling (TUNEL) assay
To determine cell apoptosis, the TUNEL Assay Kit (In Situ Cell Death Detection Kit, POD) (Sigma-Aldrich, 11684817910) based on TUNEL assay was used according to the manufacturer’s protocol. Nuclear staining was carried out using Hoechst (Thermo Fisher 33342) diluted at a 1:10,000 ratio. After incubating the cells for 10 min, the staining solution was removed, and the cells were washed three times with PBS. The stained cells were then observed under a fluorescence microscope (EVOS FL Auto, Thermo Fisher Scientific, Waltham, MA, USA).
Western blotting analysis
HNC cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Invitrogen, IBS-BR002) containing 50 mM Tris-HCl, pH7.5, 150 mM sodium chloride, 0.5% sodium deoxycholate, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 2 mM EDTA, a protease inhibitor cocktail, and PhoSTOP (Roche Molecular Biochemicals, Bas). Protein lysate (20 µg) was separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE, 10% gel) and transferred to polyvinylidene difluoride (PVDF) membranes. After transfer, each membrane was blocked with 5% skim milk for 2 h at room temperature and washed three times with 0.1% TBST (Tris-buffered saline containing Tween-20). Following the washes, cells were incubated overnight with the primary antibody (1:1,000) at 4 °C and washed three times with 0.1% TBST. The membranes were incubated with a secondary antibody (1:5,000) for 2 h at room temperature. Protein bands were visualized using ECL reagents (GE Healthcare Life Sciences, RPN2235) and detected using an ImageQuantTM LAS 4000 (Fuji Film, Tokyo, Japan).
Induction and inhibition of necroptosis
Necroptosis in HNC cells was induced using TCZ (TNF-α + cycloheximide + zVAD-fmk) [41], a combination of TNF-α (30 µg/mL), CHX (300 µM), and zVAD-fmk (10 µM), while necroptosis was inhibited using Nec-1 (50 µM), GSK872 (10 µM), or NSA (2 µM). Cells were pretreated with necroptosis inhibitors for 1 h, followed by treatment with either LP or TCZ. To identify the molecules involved in LP-induced necroptosis, cells were treated with NAC, MnTBAP, or a reactive nitrogen species (RNS) scavenger along with LP.
Immunocytochemistry microscopy
HNC cells were cultured on coverslips to 80% confluence and treated with LP for 12 h. After treatment, cells were washed with PBS and fixed with 4% paraformaldehyde for 15 min. Blocking was performed with 10% bovine serum albumin (BSA) containing 0.1% Triton X-100 (Sigma-Aldrich, 93443) for 1 h. Cells were then incubated overnight at 4 °C with a primary antibody solution. After washing with PBS, a secondary antibody (Alexa Fluor 488-conjugated goat anti-rabbit, A11034) was applied and incubated at room temperature for 1 h. Finally, the nuclei were stained with Hoechst (diluted 1:1,000) for 10 min, washed three times with PBS, and visualized under a fluorescence microscope (EVOS FL Auto, Thermo Fisher Scientific, Waltham, MA, USA).
RNA interference analysis
HNC cells were transfected with 500 pmol of double-stranded siRNAs targeting human RIP3, MLKL, or a negative control, synthesized by Bioneer (Deajeon, Korea), using Lipofectamine RNAi MAX (Invitrogen, 13778150) according to the manufacturer’s protocols, and 24 h post-transfection, the cells were exposed to LP, followed by protein collection at the indicated time points for subsequent experiments.
Specific siRNA sequences were as follows: for RIP3 (sense: 5’- CAG CAA UAG GAG AUU UUC U -3’; antisense: 5’- AGA AAA UCU CCU AUU GCU G -3’), for MLKL (sense: 5’- GAC GUG AAC AGG AAG CUG A -3’; antisense: 5’- UCA GCU UCC UGU UCA CGU C -3’), and for the negative control siRNA (sense: 5’- UUC UCC GAA CGU GUC ACG UTT -3’; antisense: 5’- ACG UGA CAC GUU CGG AGA ATT -3’).
JC-1, MitoRed, and MitoSOX staining
HNC cells were seeded at 1 × 105 cells/well in a 12-well plate and incubated for 24 h. After treatment with LP for 6 h, the culture medium was removed, and the cells were washed with PBS. JC-1 (Thermo Fisher, D-1168), MitoRed (Sigma-Aldrich, 53271), and MitoSOX (Thermo Fisher, M36008) were each diluted 1:1,000 in fresh culture medium and applied to the cells. The cells were incubated at 37 °C for 30 min, followed by PBS washing. Living cells were observed under a fluorescence microscope (EVOS FL Auto, Thermo Fisher, Waltham, MA, USA).
Enzyme-linked immunosorbent assay (ELISA)
FaDu, SCCQLL1, and SNU1041 cells were seeded into six-well plates and LP-treated for 12 h. After treatment, the supernatants were collected and centrifuged at 1,300 rpm for 10 min to remove cell debris. The Human IL-6 ELISA Kit (Thermo Fisher, Cat # BMS213HS) was used to detect the concentration of the indicated molecules following the manufacturer’s instructions.
Statistical analysis
Statistical comparisons between groups were carried out using the non-opposite t-test, and differences of < 0.05 (*), < 0.01(**), and < 0.001(***) were considered significant. All experiments were conducted at least three times.
Ethical compliance statement
This study did not involve human primary cells or animal experiments, and therefore Institutional Review Board (IRB) approval and ethical compliance with human or animal studies were not applicable.
| Results | ▴Top |
Treatment of LP induces HNC cell death
To investigate the effect of LP on cancer cells, we cultured HNC cells in plasma-treated medium and counted the number of cells. The plasma-treated medium was prepared by treating the cell culture media with NTP, plasma with N2 at 200 sccm and Ar at 1,500 sccm, for 2 min (Fig. 1a). After culturing the HNC cells in LP, we observed a decrease in the cell count of several HNC cell lines (Fig. 1b). We conducted a quantitative analysis and found that the cell numbers decreased as the exposure time to LP increased. A cell viability test was conducted using various doses of NTP to investigate the dose-dependent effects of NTP. Cell viability significantly decreased when the cells were treated with higher doses of NTP (Fig. 1c, d). On the other hand, treatment of LP in normal cells does not affect cell viability (Supplementary Material 1, wjon.elmerpub.com). In addition, LP treatment in the 3D spheroid culture of FaDu cells decreased the spheroid size (Fig. 1e). Cell viability of the spheroid culture also decreased by LP treatment (Supplementary Material 2A, wjon.elmerpub.com), and it was similar to that of the LP-treated two-dimensional (2D) culture of FaDu cells. The necroptosis markers (p-RIP1, p-RIP3, and p-MLKL) were expressed in LP-treated spheroid culture cells (Supplementary Material 2B, wjon.elmerpub.com). Next, we conducted a TUNEL assay to determine whether cancer cell death by LP occurred owing to apoptosis. We observed no TUNEL positive cells in FaDu and SNU1041 cells exposed to LP, suggesting that LP caused non-apoptotic cancer cell death (Fig. 1f, g).
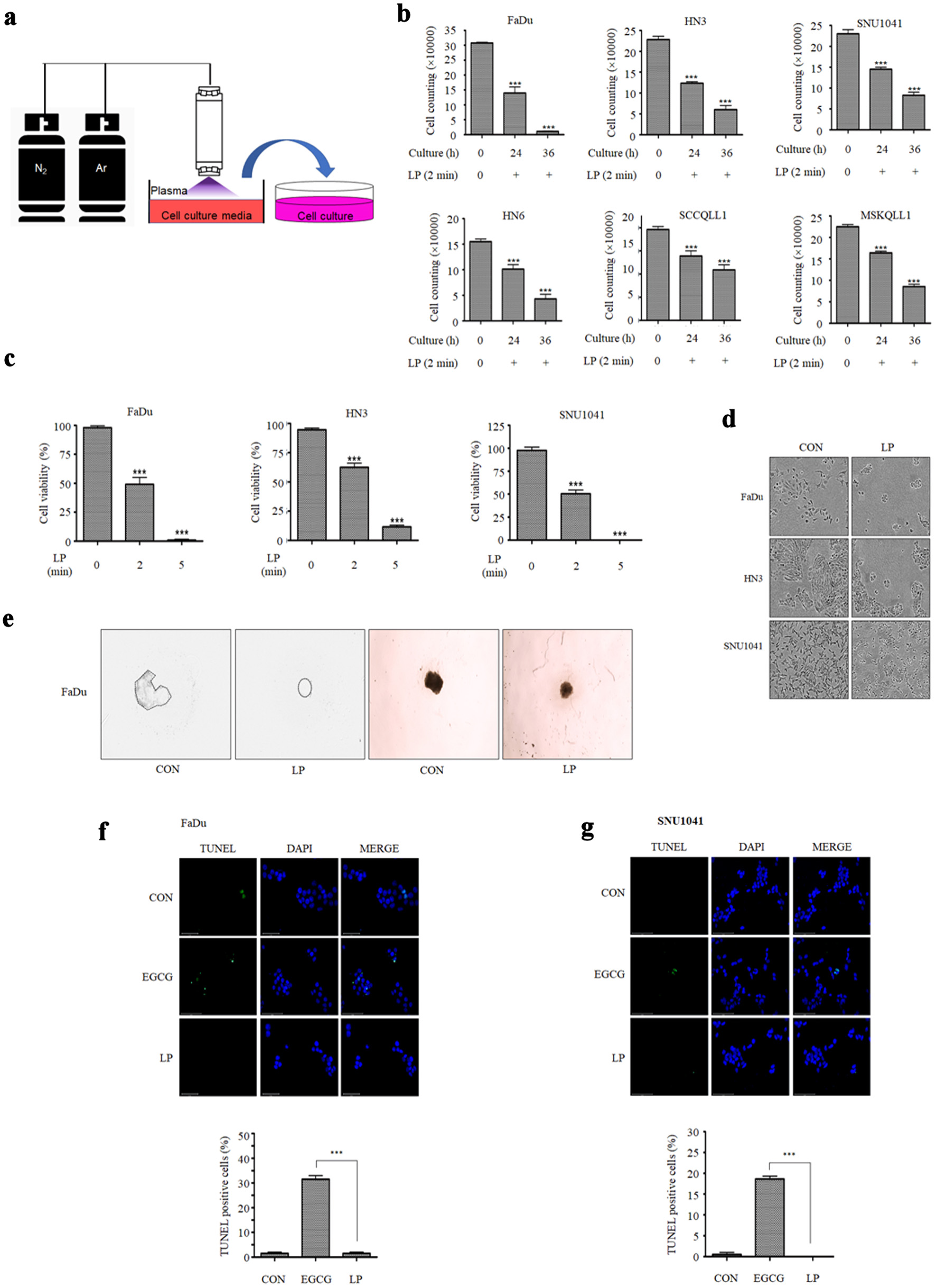 Click for large image | Figure 1. Liquid plasma (LP) induces cell death in HNC cells. (a) N2 gas was set to 200 standard cubic centimeters per minute (sccm) and Ar gas to 1,500 sccm. Cell culture medium was treated with N2/Ar plasma for 2 min per milliliter. (b) HNC cells were treated with LP for 24 or 36 h, stained with trypan blue, and then number of live cells was measured. (c) FaDu, HN3, and SNU1041 cells were treated with LP (N2/Ar plasma-treated medium for 2 min or 5 min per milliliter), and the viability was measured using CCK-8 assays after 24 h. (d) The morphology of cells was observed under a microscope 24 h after LP treatment. (e) A 3D spheroid culture of FaDu cells was observed following LP treatment. (f) FaDu cells and (g) SNU1041 cells were subjected to TUNEL assays (scale bar: 50 µm). *P < 0.05, **P < 0.01, ***P < 0.001. HNC: head and neck cancer; CCK: Cell Counting Kit; 3D: three-dimensional. |
LP induces necroptotic cell death in HNC cells
LP induces non-apoptotic programmed cell death in HNC (Fig. 1f, g). Therefore, we investigated whether it could cause necroptosis, which is one of the forms of the non-apoptotic cell death, in HNC cells. The necroptosis markers, p-RIP1, p-RIP3, and p-MLKL, increased in FaDu and SNU1041 cells treated with LP for 6 or 12 h. (Fig. 2a). The expression of p-MLKL was induced, and it was localized in the plasma membrane by LP or TCZ treatment (Supplementary Material 3, wjon.elmerpub.com). The expression of p-RIP3 was also high in LP-treated FaDu cells in immunofluorescence microscopy (Fig. 2b). We treated HNC cells with LP with or without a necroptosis inhibitor (Nec-1, GSK872, and necrosulfonamide (NSA)) to determine whether LP-induced cancer cell death is caused by the RIP3-MLKL signaling pathway. Upon treatment of cells with LP and a necroptosis inhibitor, cell viability significantly increased compared with those treated with LP alone (Fig. 2c). Western blot results also showed that necroptosis markers decreased after treatment with LP and necroptosis inhibitors (Fig. 2d). Cells were treated with LP upon RIP3 and MLKL knockdown to confirm whether LP can cause necroptosis. Upon knockdown of RIP3 expression in FaDu cells with siRIP3 RNA and subsequent treatment with LP, necroptosis marker levels decreased, and cell viability increased compared with those groups not treated with siRIP3. (Fig. 3a, c, d). Similar results were obtained when MLKL was knocked down with siMLKL RNA (Fig. 3b, e, f). We also confirmed that the amount of MLKL oligomers increased upon treatment with LP (Fig. 3g). These results indicate that necroptosis occurred when cancer cells were treated with LP.
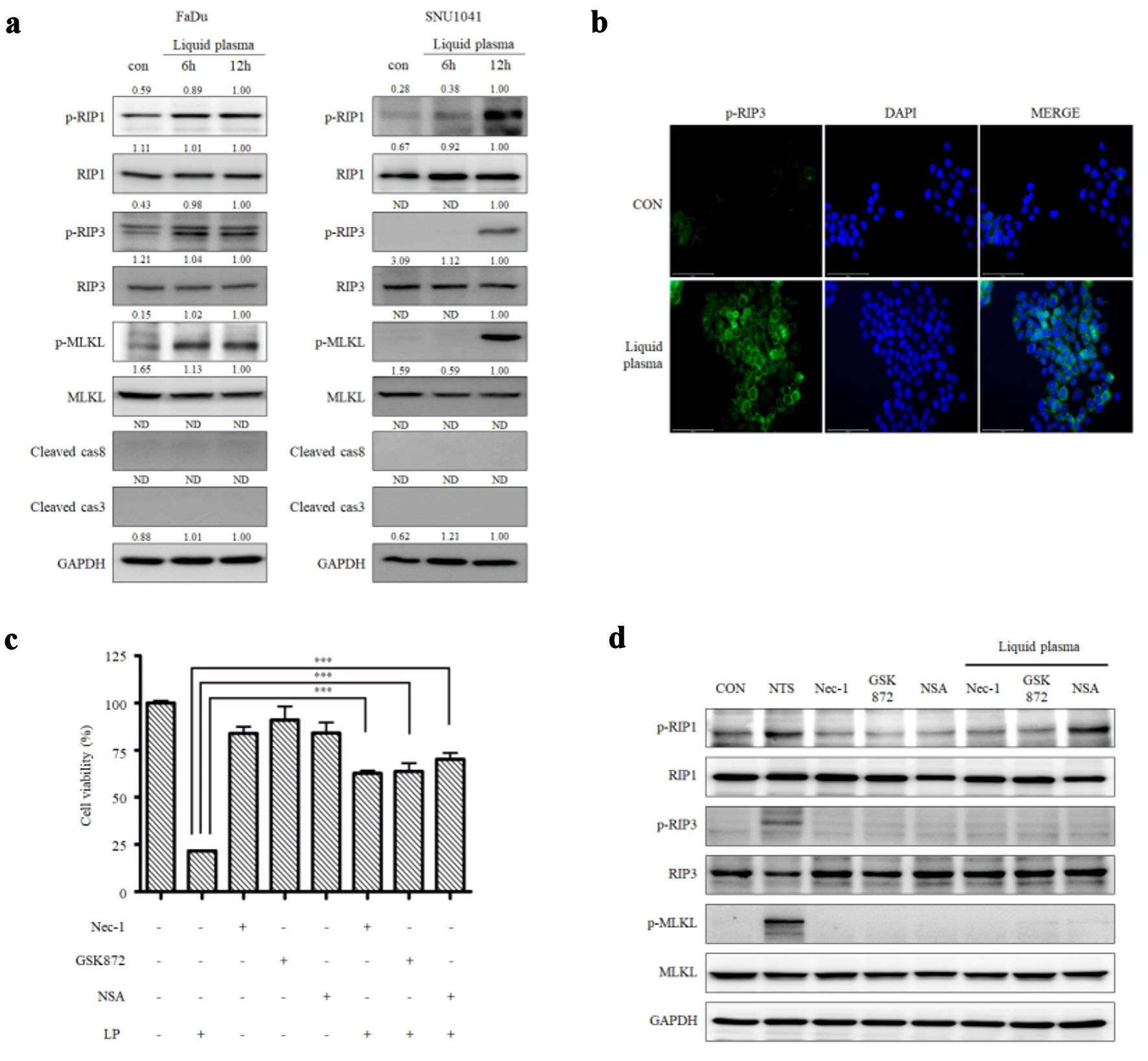 Click for large image | Figure 2. Liquid plasma (LP) induces necroptosis in FaDu and SNU1041 cells. (a) FaDu and SNU1041 cells were treated with LP for 6 or 12 h, and the expression levels of p-RIP1, RIP3, p-RIP1, RIP, p-MLKL, MLKL, cleaved caspase-8, cleaved caspase-3, and GAPDH were analyzed by Western blotting. (b) FaDu cells were treated with LP for 6 h, and p-RIP3 expression was visualized using fluorescence staining (scale bar: 50 µm). (c) FaDu cells were treated with LP, a p-RIP1 inhibitor (Nec-1), a p-RIP3 inhibitor (GSK872), and a p-MLKL inhibitor (NSA), and cell viability was measured using CCK-8 assays. (d) The expression levels of p-RIP1, RIP3, p-RIP1, RIP, p-MLKL, MLKL, and GAPDH were analyzed by Western blotting. *P < 0.05, **P < 0.01, ***P < 0.001. RIP: receptor-interacting protein; Nec-1: necrostatin-1; MLKL: mixed lineage kinase domain-like protein; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; NSA: necrosulfonamide; CCK: Cell Counting Kit. |
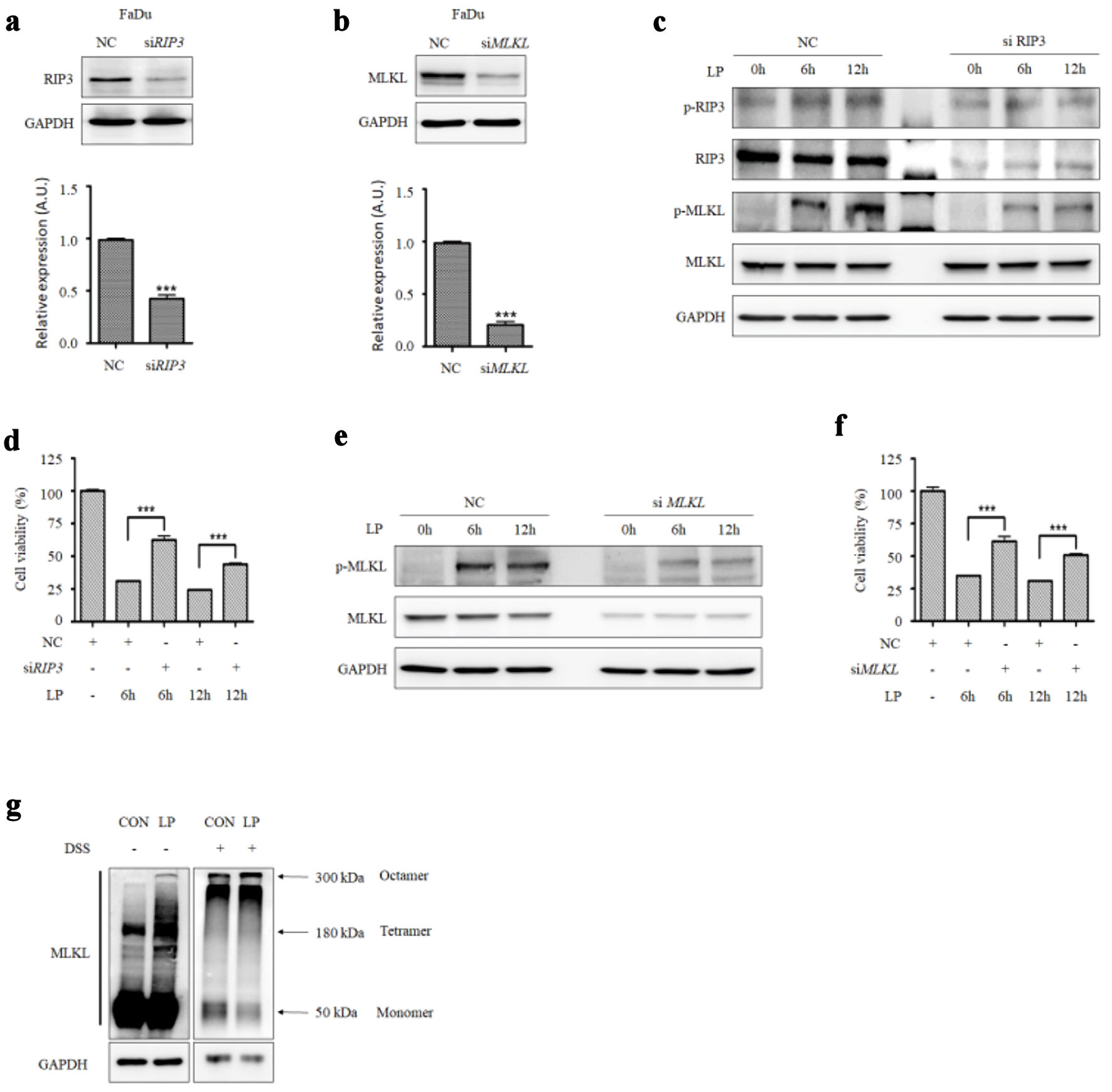 Click for large image | Figure 3. RIP3 and MLKL levels affect the sensitivity to liquid plasma (LP) in HNC cells. (a) RIP3 and (b) MLKL were knocked down in FaDu cells using siRNA. (c) FaDu cells transfected with siRIP3 RNA and (e) siMLKL RNA were treated with LP for 6 or 12 h, and protein expression levels (RIP3, MLKL, and their phosphorylated forms) were analyzed by Western blotting. (d) FaDu cells transfected with siRIP3 RNA and (f) siMLKL RNA were treated with LP for 6 or 12 h, and cell viability was measured using Cell Counting Kit (CCK-8) assays. (g) MLKL oligomerization was detected by Western blotting under non-reducing conditions. *P < 0.05, **P < 0.01, ***P < 0.001. HNC: head and neck cancer; RIP: receptor-interacting protein; MLKL: mixed lineage kinase domain-like protein; GAPDH: glyceraldehyde-3-phosphate dehydrogenase. |
LP induces necroptosis of HNC cells via peroxynitrite
Distilled water was treated with NTP to generate LP, which was then analyzed using a chromatographic ion analyzer to determine which substance in LP causes necroptosis. We found that RNS, nitrite and nitrate were present in the LP (Fig. 4a). Next, to determine which RNS (NO, NO2, or NO3) was the main effector, fibroblasts and FaDu cells were treated with LP, DetaNO, NaNO2, and NaNO3. NaNO2 had no effect on fibroblast and FaDu cell viability; however, DetaNO and NaNO3 treatment decreased cell viability as the dose increased (Fig. 4b, c). Western blot analysis showed that p-MLKL was expressed when LP and 50 mM NaNO3 were treated, and the expression of cleaved caspase 3 and 9 increased in the 0.5 mM DetaNO-treated group (Fig. 4d). These results suggest that LP and NO3 induce necroptosis, and NO induces apoptosis. In addition, when the peroxynitrite (ONOO-) inhibitor, which is MnTBAP and non-cytotoxic (Supplementary Material 4, wjon.elmerpub.com), was simultaneously treated with LP, LP treatment did not induce cancer cell death, suggesting that a main component of LP for necroptosis induction is ONOO- (Fig. 4e, f). The increased expression of p-RIP3 in the group treated with LP was observed through immunofluorescence microscopy, and treatment of the ONOO- inhibitor decreased pRIP3, which is induced by LP (Fig. 4g, h). Based on these results, we conclude that LP causes cancer cell death via ONOO-.
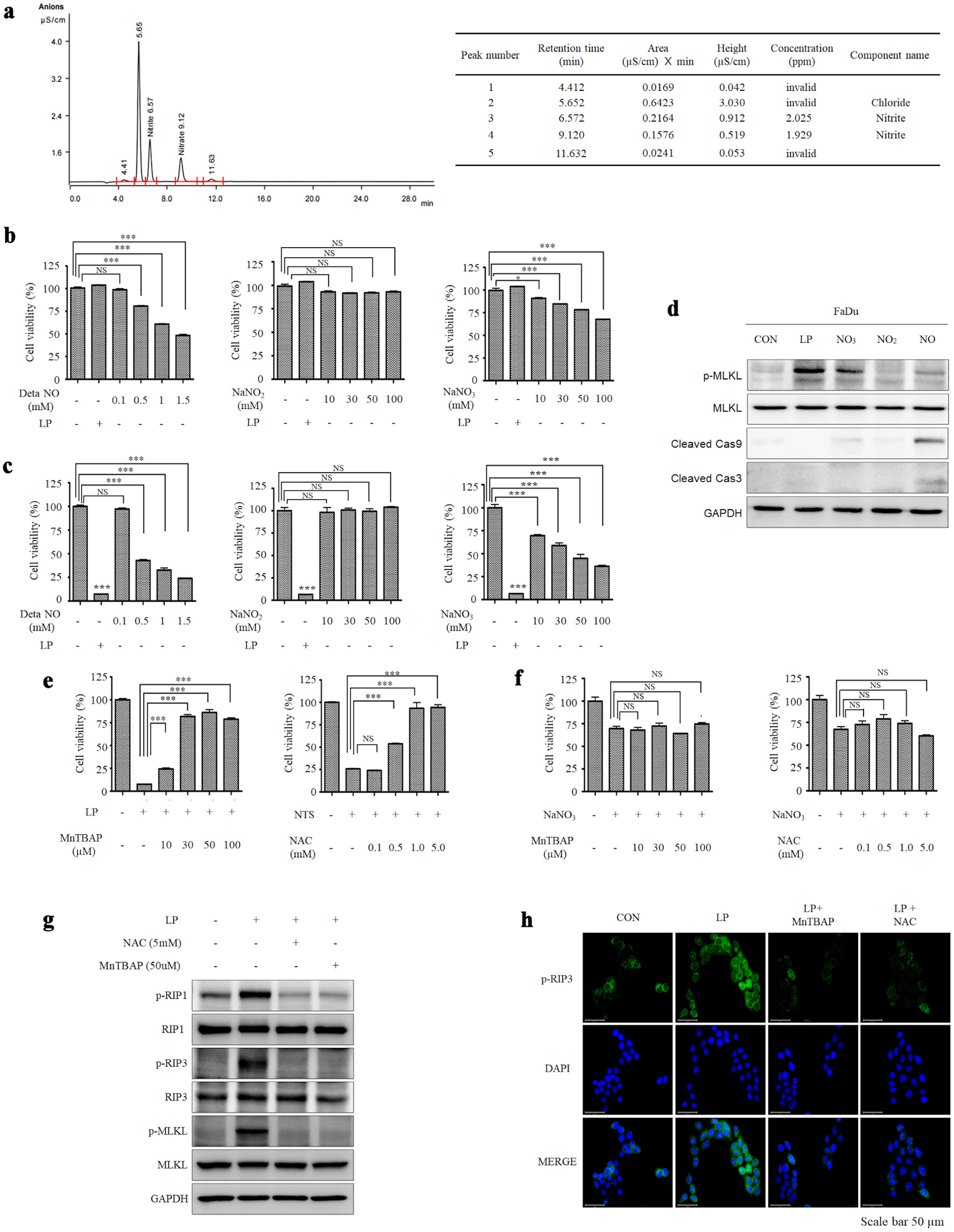 Click for large image | Figure 4. Peroxynitrite in liquid plasma (LP) induces necroptosis in HNC cells. (a) The components of LP were analyzed using a chromatographic ion analyzer. (b) Human dermal fibroblasts (HDF) and (c) FaDu cells were treated with DetaNO, NaNO2, NaNO3, and LP, representing nitrite and nitrate, which are presumed components of plasma. (d) FaDu cells were treated with DetaNO, NaNO2, NaNO3, and LP, and the expression levels of p-MLKL, MLKL, cleaved caspase-9, cleaved caspase-3, and GAPDH were analyzed by Western blotting. (e) FaDu cells were treated with LP, and the peroxynitrite inhibitor MnTBAP was added at different concentrations. Cell viability was measured using CCK-8 assays. (f) FaDu cells were treated with NaNO3, and the peroxynitrite inhibitor MnTBAP was added at different concentrations. Cell viability was measured using CCK-8 assays. (g) FaDu cells were treated with NAC and MnTBAP for 12 h, and the expression levels of p-RIP2, RIP1, p-RIP3, RIP3, p-MLKL, MLKL, and GAPDH were analyzed by Western blotting. (h) The expression of p-RIP3 following LP treatment was confirmed using fluorescence staining (scale bar: 50 µm). *P < 0.05, **P < 0.01, ***P < 0.001. HNC: head and neck cancer; MLKL: mixed lineage kinase domain-like protein; MnTBAP: manganese (III) tetrakis (4-benzoic acid) porphyrin chloride; RIP: receptor-interacting protein; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; DAPI: 4′,6-diamidino-2-phenylindole; NS: non-significant. |
LP treatment increases mitochondrial reactive oxygen species (ROS) in HNC cells
NTP affects the intercellular ROS and increases mitochondrial stress in cancer. Therefore, effect of LP on the mitochondrial complex were examined. When cells were treated with LP for 0, 6, or 12 h, no difference in the expression of each mitochondrial complex component in FaDu cells was observed, as shown in Western blots. (Fig. 5a). However, among the mitochondrial stress-involved proteins, a decrease in MAVS expression was observed (Fig. 5b). We observed no change in the number of mitochondria and membrane potential following LP treatment by MitoRed and JC-1 staining experiments, respectively (Fig. 5c, d). However, MitoSOX staining revealed an increase in mitochondrial ROS levels by LP treatment (Fig. 5e).
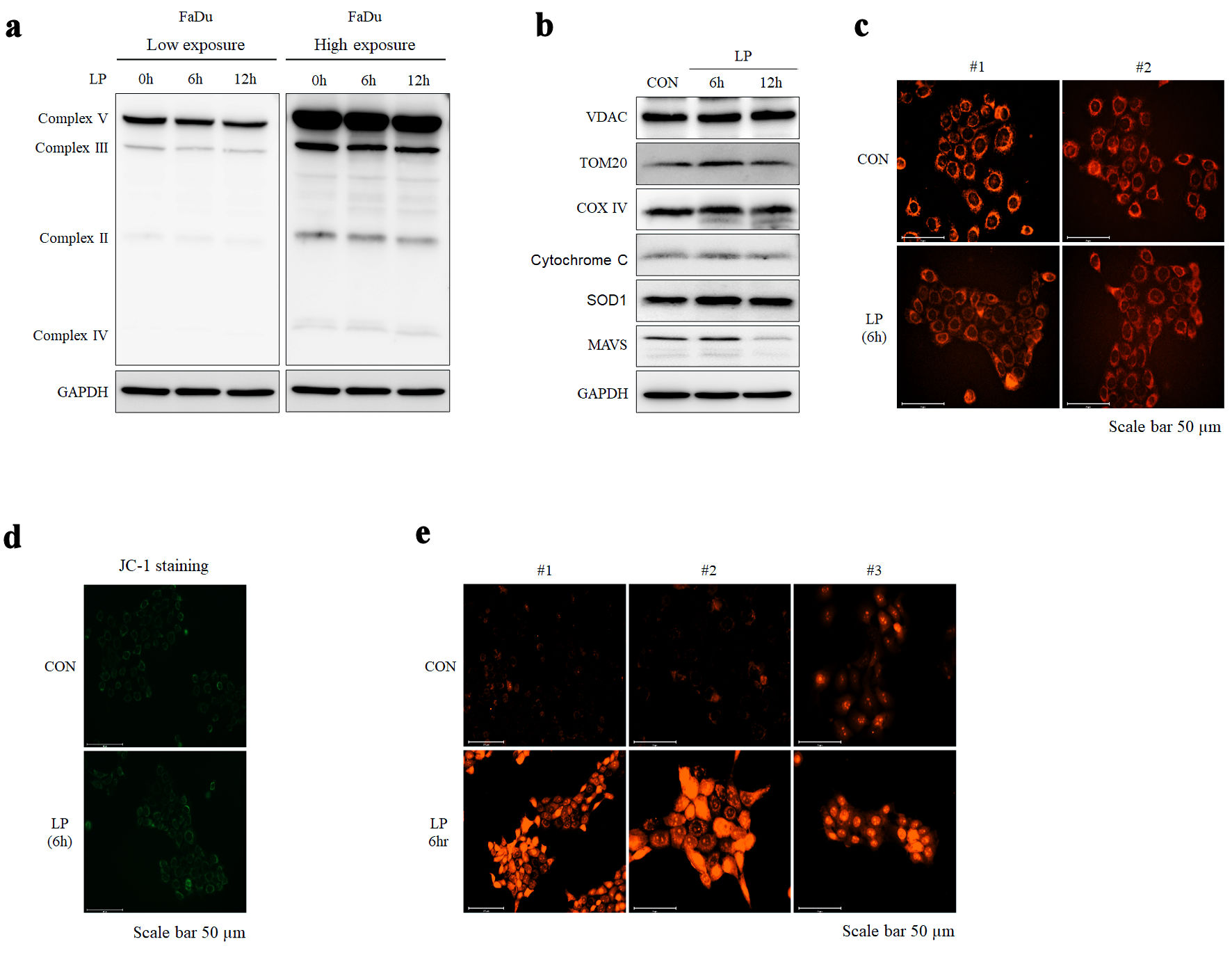 Click for large image | Figure 5. Liquid plasma (LP) treatment induces mitochondrial stress in HNC cells. (a, b) The expression of mitochondria complex and mitochondrial proteins (VADC, TOM20, COX IV, cytochrome c, SOD, MAVS) following LP treatment was confirmed using Western blotting. (c, d) Changes in mitochondrial morphology after LP treatment were observed using fluorescence staining with MitoRed and JC-1, respectively (scale bar: 50 µm). (e) ROS levels following LP treatment were assessed using fluorescence staining with MitoSox (scale bar: 50 µm). HNC: head and neck cancer; MAVS: mitochondrial antiviral signaling; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; ROS: reactive oxygen species. |
LP treatment decreases necroptosis-induced inflammation through degrading MAVS
Aggregation of MAVS protein activates an inflammatory response via NF-κB. Therefore, we hypothesized that LP is involved in the inflammatory response by reducing MAVS. In general, p-NF-κB and MAVS levels increase during necroptosis, which was confirmed by treatment with TCZ, a drug that induces necroptosis, in FaDu cells (Fig. 6a). In contrast, upon treatment of HNC cell lines with LP, a decrease in MAVS and p-NF-κB was observed (Fig. 6b). Western blotting was performed after DSS treatment to determine whether LP treatment reduced MAVS aggregation. MAVS aggregation was reduced in FaDu cells transfected with MAVS. We obtained the same results when we transfected MAVS-negative 293T cells with MAVS (Fig. 6c). Immunofluorescence microscopy revealed that LP treatment reduced the NF-κB level, compared to TCZ treatment (Fig. 6d). When interleukin (IL)-6, an inflammatory cytokine, was measured by ELISA, IL-6 significantly decreased upon LP treatment (Fig. 6e). These results indicate that LP treatment induces necroptosis but inhibits inflammatory responses.
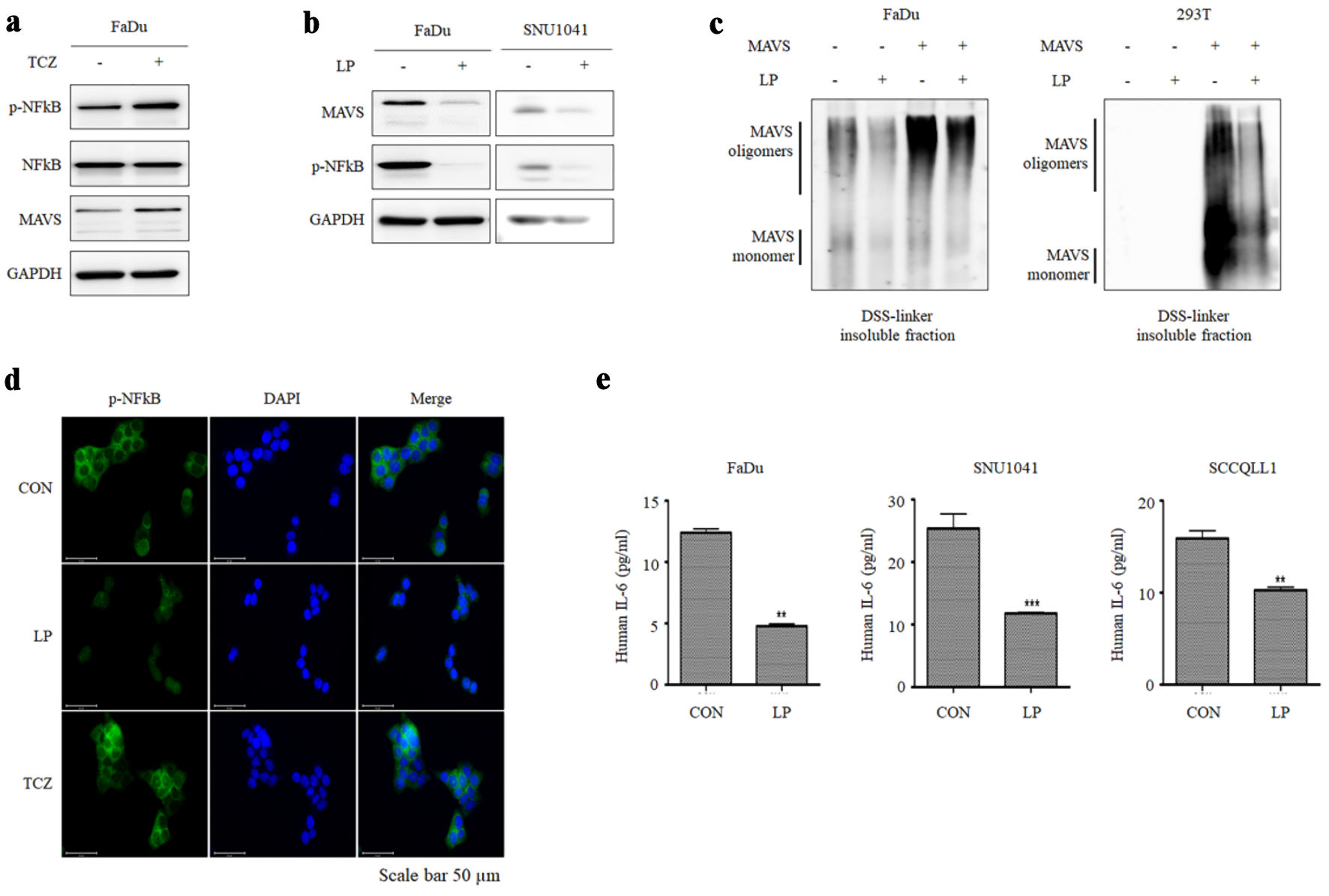 Click for large image | Figure 6. Liquid plasma (LP) inhibits necroptosis-mediated inflammation by degrading MAVS aggregates. (a) FaDu cells were treated with TCZ (30 µg/mL TNF-α, 300 µM CHX, and 10 µM zVAD-fmk) for 12 h, and the expression of MAVS and p-NF-κB was observed. (b) LP treatment suppressed the expression of MAVS and p-NF-κB in FaDu and SNU1041 cells. (c) After inducing MAVS oligomerization in FaDu and 293T cells, the degradation of oligomerized MAVS by LP treatment was observed. (d) FaDu cells treated with both LP and TCZ, changes in NF-κB expression levels and nuclear translocation were analyzed using fluorescence microscopy (scale bar: 50 µm). (e) LP treatment inhibited TCZ-induced IL-6 production in FaDu and SNU1041 cells. IL-6 levels in the supernatant were measured using ELISA assay. **P < 0.01, ***P < 0.001. MAVS: mitochondrial antiviral signaling; TNF-α: tumor necrosis factor alpha; CHX: cycloheximide; NF-κB: nuclear factor kappa B; IL: interleukin; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; DAPI: 4′,6-diamidino-2-phenylindole; TCZ: TNF-α + cycloheximide + zVAD-fmk. |
| Discussion | ▴Top |
Necroptosis is a regulated form of cell death involved in cancer immunosurveillance through both innate and adaptive immunity. Necroptosis plays a role in innate immunity as it affects cytokine secretion by dendritic cells, promoting the anti-tumor immune response of natural killer T cells, and maintaining homeostasis in peripheral T cells. Adaptive immunity is activated as phagocytic cells secrete pro-inflammatory cytokines in the damage associated molecular pattern caused by necroptosis [42]. However, the proinflammatory molecules secreted during necroptosis, such as IL-1α, can promote cancer cell proliferation, foster angiogenesis, and accelerate metastasis, contributing to tumor progression [43, 44]. Thus, inducing necroptosis without triggering inflammation is critical for its therapeutic application.
RONS generated by plasma have been widely applied in medicine, particularly for their selective ability to kill cancer cells, making plasma a promising cancer therapeutic tool [45, 46]. The composition and effects of plasma-generated RONS, including O, O3, •OH, H2O2, O2•-/•OOH, •NO, ONOO-, OONOO-, NO2-, and NO3-, vary depending on the gas supply and generation system [47]. Among the various RONS generated by plasma, NO plays a dual role. Although NO is important for wound healing and tissue repair [48], it also exerts a pro-apoptotic effect by inhibiting ribonucleotide reductase, leading to DNA damage and cell death [49]. Additionally, nitric oxide synthase (NOS)/NO promotes apoptosis by inhibiting autophagy in liver cancer cells [50] and induces apoptosis in oral squamous cell carcinoma cells by facilitating p53 accumulation [51]. In this study, treatment of HNC cells with DetaNO confirmed that apoptosis was induced, as evidenced by increased levels of cleaved caspase-3. In contrast, treatment of HNC cells with NaNO3 led to necroptosis, marked by cell death and increased expression of p-RIP3, mirroring the effects observed with LP treatment. These results suggest that different nitrogen species distinctly influence the mode of cell death, with NO driving apoptosis and NO3- contributing to necroptosis. Among the various RONS generated by plasma, Xu et al identified ONOO- as a key mediator of plasma-induced cancer cell death [52]. Based on these findings, we speculate that ONOO- plays a pivotal role in LP-induced necroptosis.
Previous studies have demonstrated that the type of cell death induced by plasma treatment depends on various factors, including the gas composition and molecular context of the target cell [53]. When necroptosis occurs, NF-κB is activated, causing an inflammatory response. The downstream signaling caused by the activation of NF-κB induces cancer proliferation, which poses some risks for the application of necroptosis to cancer [8, 43]. However, in this study, we demonstrated that LP-induced necroptosis, unlike normal necroptosis, suppressed MAVS expression in the mitochondria, resulting in decreased NF-κB activation and IL-6 expression.
MAVS is known to undergo ubiquitin-proteasome-mediated degradation in resting cells to maintain immune homeostasis [54]. Multiple E3 ligases, including MARCH5, Smurf1/2, AIP4, VHL, RNF5, RNF115, and RNF125 mediate K48-linked ubiquitination and proteasomal degradation of MAVS to regulate immune responses after viral infection [55-61]. Among these, our previous study demonstrated that MARCH5 resolves MAVS aggregates during antiviral signaling and prevents excessive immune activation [55]. RONS generated by plasma are well-known to modulate cellular signaling pathways through oxidative stress and K48-linked ubiquitination and proteasomal degradation [62]. In our previous work, LP-derived RONS were shown to induce the ubiquitination of mTOR via RNF126 in leukemia cells [63], as well as HSPA5 [28] and AKT [64] via MUL1 E3 ligase in HNC cells. These findings suggest that plasma-derived RONS may similarly regulate MAVS ubiquitination, contributing to the suppression of inflammatory responses during necroptosis. However, further studies are required to identify the specific E3 ligases involved in MAVS ubiquitination under LP treatment and to elucidate their role in modulating inflammatory responses during necroptosis.
Future studies should investigate the broader implications of LP’s anti-inflammatory activity in various cancer types and its interaction with the immune system within the tumor microenvironment. While LP’s ability to suppress inflammation in HNC cells is promising, future studies should investigate whether this effect is universal across various cancer types. Moreover, detailed exploration of how LP modulates immune cell dynamics, such as T-cell activation or macrophage polarization, could provide new insights into its role in the tumor microenvironment and enhance its therapeutic potential. Necroptosis after irradiation promotes tumor cell repopulation, which is the major reason for radiotherapy resistance and recurrence due to necroptosis-induced inflammation [65]. Necroptosis is involved in the pathogenesis of many inflammatory disorders. Therefore, it must be induced without inflammation. In this study, we demonstrated that LP could induce necroptosis in HNC cells without inflammatory responses through the degradation of MAVS.
While our findings indicate that LP treatment increases mitochondrial ROS levels without causing detectable mitochondrial damage, the precise mechanisms underlying this phenomenon remain unclear. Previous studies suggest that specific alterations in the electron transport chain, such as reverse electron transfer through complex I, may lead to enhanced superoxide generation without affecting mitochondrial membrane potential [66, 67]. However, the potential contribution of non-mitochondrial sources of ROS, such as the cytoplasm, cannot be ruled out. Future studies are needed to elucidate these mechanisms and to explore how this ROS production influences the therapeutic efficacy and safety of LP in cancer treatment.
Our previous findings demonstrated that NTP induces selective apoptosis in cancer cells by downregulating the NRF2-induced ROS scavenger system and inhibiting autophagy, while sparing normal cells through enhanced antioxidant gene expression and autophagic flux [68]. In contrast, the current study shows that LP primarily induces necroptosis in HNC cells, with ONOO- identified as a key effector. This transition from apoptosis to necroptosis highlights the versatility of plasma-based therapies in modulating RCD. However, the mechanisms driving this selective induction of necroptosis in cancer cells, while sparing normal cells, remain unclear and require further investigation. As these findings are based on in vitro experiments, in vivo studies are crucial to confirm the therapeutic efficacy and safety of LP under physiological conditions. Therefore, it might be necessary to investigate whether LP can induce necroptosis in a mouse xenograft model using FaDu cells, as well as determine the expression levels of proinflammatory cytokines and the degradation of MAVS in a tumor mass. It is also essential for future research to identify the E3 ligase that is mediated in LP-induced necroptosis so that the mechanisms of LP-induced cancer cell death are clarified. In addition, it has not been identified how ONOO- can induce necroptosis. Thus, it is necessary to determine the role of ONOO- in necroptosis induction to understand how LP induces necroptosis without inflammation in HNC cells. Furthermore, the development of effective delivery systems, such as injectable formulations or localized applications, is necessary to enable its clinical translation. Despite these challenges, LP’s ability to induce necroptosis without triggering inflammatory responses underscores its potential as a promising therapeutic strategy for apoptosis-resistant cancers and necroptosis-related neurodegenerative and inflammatory diseases.
Conclusions
LP induces non-inflammatory necroptosis in HNC cells, offering a potential therapeutic strategy to eliminate cancer cells without triggering necroptosis-mediated inflammation or tumor recurrence. These findings suggest LP as a promising treatment for drug-induced apoptosis-resistant cancers, as it induces necroptosis instead of apoptosis while minimizing the risks of inflammatory disease and tumor repopulation.
| Supplementary Material | ▴Top |
Suppl 1. LP does not affect normal cell viability.
Suppl 2. Cell viability and necroptosis marker expression in FaDu cells.
Suppl 3. pMLKL expression and localization was visualized by immunofluorescence microscopy.
Suppl 4. MnTBAP does not affect FaDu cell viability.
Acknowledgments
The authors thank all the staff supporting this study.
Financial Disclosure
This work was supported by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HR21C1003 and RS-2024-00438448), and R&D project for treatment of infectious medical waste, funded by the Ministry of Environment (MOE) of the Republic of Korea (2021003350001).
Conflict of Interest
The authors declare no competing interest.
Informed Consent
Not applicable.
Author Contributions
JHC performed and analyzed the experiments. SK analyzed the experimental results and wrote the manuscript. YSL analyzed the experimental results and wrote the manuscript. YSY designed and analyzed the experiments. JYJ analyzed the experimental results and edited the manuscript. YSS analyzed the experimental results and edited the manuscript. C-HK designed and analyzed experiments, wrote manuscript, and supervised this study. All authors reviewed and commented on the manuscript.
Data Availability
The datasets generated and/or analyzed during the study are available from the corresponding author upon reasonable request.
| References | ▴Top |
- Lockshin RA, Williams CM. Programmed Cell Death—I. Cytology of degeneration in the intersegmental muscles of the pernyi silkmoth. J Insect Physiol. 1965;11:123-133.
doi pubmed - Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347-364.
doi pubmed - Inoue H, Tani K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. 2014;21(1):39-49.
doi pubmed - Dhuriya YK, Sharma D. Necroptosis: a regulated inflammatory mode of cell death. J Neuroinflammation. 2018;15(1):199.
doi pubmed - Hanna-Addams S, Liu S, Liu H, Chen S, Wang Z. CK1alpha, CK1delta, and CK1epsilon are necrosome components which phosphorylate serine 227 of human RIPK3 to activate necroptosis. Proc Natl Acad Sci U S A. 2020;117(4):1962-1970.
doi pubmed - Zhou Y, Xiang Y, Liu S, Li C, Dong J, Kong X, Ji X, et al. RIPK3 signaling and its role in regulated cell death and diseases. Cell Death Discov. 2024;10(1):200.
doi pubmed - Makin G, Dive C. Apoptosis and cancer chemotherapy. Trends Cell Biol. 2001;11(11):S22-26.
doi pubmed - Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714-726.
doi pubmed - Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo J, et al. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther. 2007;6(5):1641-1649.
doi pubmed - Oliver Metzig M, Fuchs D, Tagscherer KE, Grone HJ, Schirmacher P, Roth W. Inhibition of caspases primes colon cancer cells for 5-fluorouracil-induced TNF-alpha-dependent necroptosis driven by RIP1 kinase and NF-kappaB. Oncogene. 2016;35(26):3399-3409.
doi pubmed - McCabe KE, Bacos K, Lu D, Delaney JR, Axelrod J, Potter MD, Vamos M, et al. Triggering necroptosis in cisplatin and IAP antagonist-resistant ovarian carcinoma. Cell Death Dis. 2014;5(10):e1496.
doi pubmed - Bonapace L, Bornhauser BC, Schmitz M, Cario G, Ziegler U, Niggli FK, Schafer BW, et al. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J Clin Invest. 2010;120(4):1310-1323.
doi pubmed - Kong MG, Kroesen G, Morfill G, Nosenko T, Shimizu T, van Dijk J, Zimmermann JL. Plasma medicine: an introductory review. New Journal of Physics. 2009;11.
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579-591.
doi pubmed - Tanaka H, Mizuno M, Ishikawa K, Nakamura K, Kajiyama H, Kano H, Kikkawa F, et al. Plasma-activated medium selectively kills glioblastoma brain tumor cells by down-regulating a survival signaling molecule, AKT kinase. Plasma Medicine. 2011;1:365-277.
- Zucker SN, Zirnheld J, Bagati A, DiSanto TM, Des Soye B, Wawrzyniak JA, Etemadi K, et al. Preferential induction of apoptotic cell death in melanoma cells as compared with normal keratinocytes using a non-thermal plasma torch. Cancer Biol Ther. 2012;13(13):1299-1306.
doi pubmed - Liu Y, Tan S, Zhang H, Kong X, Ding L, Shen J, Lan Y, et al. Selective effects of non-thermal atmospheric plasma on triple-negative breast normal and carcinoma cells through different cell signaling pathways. Sci Rep. 2017;7(1):7980.
doi pubmed - Guerrero-Preston R, Ogawa T, Uemura M, Shumulinsky G, Valle BL, Pirini F, Ravi R, et al. Cold atmospheric plasma treatment selectively targets head and neck squamous cell carcinoma cells. Int J Mol Med. 2014;34(4):941-946.
doi pubmed - Kaushik N, Kumar N, Kim CH, Kaushik NK, Choi EH. Dielectric barrier discharge plasma efficiently delivers an apoptotic response in human monocytic lymphoma. Plasma Processes and Polymers. 2014;11:1175-1187.
- Utsumi F, Kajiyama H, Nakamura K, Tanaka H, Hori M, Kikkawa F. Selective cytotoxicity of indirect nonequilibrium atmospheric pressure plasma against ovarian clear-cell carcinoma. Springerplus. 2014;3:398.
doi pubmed - Liedtke KR, Bekeschus S, Kaeding A, Hackbarth C, Kuehn JP, Heidecke CD, von Bernstorff W, et al. Non-thermal plasma-treated solution demonstrates antitumor activity against pancreatic cancer cells in vitro and in vivo. Sci Rep. 2017;7(1):8319.
doi pubmed - Duan J, Lu X, He G. The selective effect of plasma activated medium in an in vitro co-culture of liver cancer and normal cells. Journal of Applied Physics. 2017;12.
- Canal C, Fontelo R, Hamouda I, Guillem-Marti J, Cvelbar U, Ginebra MP. Plasma-induced selectivity in bone cancer cells death. Free Radic Biol Med. 2017;110:72-80.
doi pubmed - Kim SJ, Chung TH. Cold atmospheric plasma jet-generated RONS and their selective effects on normal and carcinoma cells. Sci Rep. 2016;6:20332.
doi pubmed - Chang JW, Kang SU, Shin YS, Kim KI, Seo SJ, Yang SS, Lee JS, et al. Non-thermal atmospheric pressure plasma induces apoptosis in oral cavity squamous cell carcinoma: Involvement of DNA-damage-triggering sub-G(1) arrest via the ATM/p53 pathway. Arch Biochem Biophys. 2014;545:133-140.
doi pubmed - Biscop E, Baroen J, De Backer J, Vanden Berghe W, Smits E, Bogaerts A, Lin A. Characterization of regulated cancer cell death pathways induced by the different modalities of non-thermal plasma treatment. Cell Death Discov. 2024;10(1):416.
doi pubmed - Kang SU, Cho JH, Chang JW, et al. Characterization of regulated cancer cell death pathways induced by the different modalities of non-thermal plasma treatment. Cell Death Dis. 2014;5:e1056.
- Kim SY, Kim HJ, Kim HJ, Kim DH, Han JH, Byeon HK, Lee K, et al. HSPA5 negatively regulates lysosomal activity through ubiquitination of MUL1 in head and neck cancer. Autophagy. 2018;14(3):385-403.
doi pubmed - Yoshikawa N, Liu W, Nakamura K, Yoshida K, Ikeda Y, Tanaka H, Mizuno M, et al. Plasma-activated medium promotes autophagic cell death along with alteration of the mTOR pathway. Sci Rep. 2020;10(1):1614.
doi pubmed - Yang X, Chen G, Yu KN, Yang M, Peng S, Ma J, Qin F, et al. Cold atmospheric plasma induces GSDME-dependent pyroptotic signaling pathway via ROS generation in tumor cells. Cell Death Dis. 2020;11(4):295.
doi pubmed - Peng S, Chen G, Yu KN, Feng Y, Zhao L, Yang M, Cao W, et al. Synergism of non-thermal plasma and low concentration RSL3 triggers ferroptosis via promoting xCT lysosomal degradation through ROS/AMPK/mTOR axis in lung cancer cells. Cell Commun Signal. 2024;22(1):112.
doi pubmed - Lunov O, Zablotskii V, Churpita O, Lunova M, Jirsa M, Dejneka A, Kubinova S. Chemically different non-thermal plasmas target distinct cell death pathways. Sci Rep. 2017;7(1):600.
doi pubmed - Kang SU, Seo SJ, Kim YS, Shin YS, Koh YW, Lee CM, Yang SS, et al. Comparative effects of non-thermal atmospheric pressure plasma on migration and invasion in oral squamous cell cancer, by Gas Type. Yonsei Med J. 2017;58(2):272-281.
doi pubmed - Fritsch M, Gunther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP, Schiffmann LM, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. 2019;575(7784):683-687.
doi pubmed - Yang CY, Tseng YC, Tu YF, Kuo BJ, Hsu LC, Lien CI, Lin YS, et al. Reverse hierarchical DED assembly in the cFLIP-procaspase-8 and cFLIP-procaspase-8-FADD complexes. Nat Commun. 2024;15(1):8974.
doi pubmed - Li J, Huang S, Zeng L, Li K, Yang L, Gao S, Guan C, et al. Necroptosis in head and neck squamous cell carcinoma: characterization of clinicopathological relevance and in vitro cell model. Cell Death Dis. 2020;11(5):391.
doi pubmed - Xu L, Davidson BJ, Murty VV, Li RG, Sacks PG, Garin-Chesa P, Schantz SP, et al. TP53 gene mutations and CCND1 gene amplification in head and neck squamous cell carcinoma cell lines. Int J Cancer. 1994;59(3):383-387.
doi pubmed - Lee SY, Park HR, Cho NH, Choi YP, Rha SY, Park SW, Kim SH. Identifying genes related to radiation resistance in oral squamous cell carcinoma cell lines. Int J Oral Maxillofac Surg. 2013;42(2):169-176.
doi pubmed - Kim SY, Chu KC, Lee HR, Lee KS, Carey TE. Establishment and characterization of nine new head and neck cancer cell lines. Acta Otolaryngol. 1997;117(5):775-784.
doi pubmed - Kang SU, Kim YS, Kim YE, Park JK, Lee YS, Kang HY, Jang JW, et al. Opposite effects of non-thermal plasma on cell migration and collagen production in keloid and normal fibroblasts. PLoS One. 2017;12(11):e0187978.
doi pubmed - Najafov A, Mookhtiar AK, Luu HS, Ordureau A, Pan H, Amin PP, Li Y, et al. TAM kinases promote necroptosis by regulating oligomerization of MLKL. Mol Cell. 2019;75(3):457-468.e454.
doi pubmed - Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K, Cheng H, et al. The role of necroptosis in cancer biology and therapy. Mol Cancer. 2019;18(1):100.
doi pubmed - Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch. 2005;446(5):475-482.
doi pubmed - Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.
doi pubmed - Sole-Marti X, Espona-Noguera A, Ginebra MP, Canal C. Plasma-conditioned liquids as anticancer therapies in vivo: current state and future directions. Cancers (Basel). 2021;13(3).
doi pubmed - Kim S, Kim CH. Applications of Plasma-Activated Liquid in the Medical Field. Biomedicines. 2021;9(11).
doi pubmed - Bruggeman PJ, Kushner MJ, Locke BR, Gardeniers JGE, Graham WG, Graves DB, Hofman-Caris RCHM, et al. Plasma-liquid interactions: a review and roadmap. Plasma Sources Science and Technology. 2016;25.
- Kang SU, Kim HJ, Ma S, Oh DY, Jang JY, Seo C, Lee YS, et al. Liquid plasma promotes angiogenesis through upregulation of endothelial nitric oxide synthase-induced extracellular matrix metabolism: potential applications of liquid plasma for vascular injuries. Cell Commun Signal. 2024;22(1):138.
doi pubmed - Murphy MP. Nitric oxide and cell death. Biochim Biophys Acta. 1999;1411(2-3):401-414.
doi pubmed - Zhang X, Jin L, Tian Z, Wang J, Yang Y, Liu J, Chen Y, et al. Nitric oxide inhibits autophagy and promotes apoptosis in hepatocellular carcinoma. Cancer Sci. 2019;110(3):1054-1063.
doi pubmed - Zhao SF, Tong XY, Zhu FD. Nitric oxide induces oral squamous cell carcinoma cells apoptosis with p53 accumulation. Oral Oncol. 2005;41(8):785-790.
doi pubmed - Xu D, Cui Q, Xu Y, Liu Z, Chen Z, Xia W, Zhang H et al. NO2- and NO3- enhance cold atmospheric plasma induced cancer cell death by generation of ONOO. AIP Advances. 2018;8:105219.
- Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106-1121.
doi pubmed - Chen Y, Shi Y, Wu J, Qi N. MAVS: A two-sided CARD mediating antiviral innate immune signaling and regulating immune homeostasis. Front Microbiol. 2021;12:744348.
doi pubmed - Yoo YS, Park YY, Kim JH, Cho H, Kim SH, Lee HS, Kim TH, et al. The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signalling. Nat Commun. 2015;6:7910.
doi pubmed - You F, Sun H, Zhou X, Sun W, Liang S, Zhai Z, Jiang Z. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat Immunol. 2009;10(12):1300-1308.
doi pubmed - Du J, Zhang D, Zhang W, Ouyang G, Wang J, Liu X, Li S, et al. pVHL Negatively Regulates Antiviral Signaling by Targeting MAVS for Proteasomal Degradation. J Immunol. 2015;195(4):1782-1790.
doi pubmed - Zhong B, Zhang Y, Tan B, Liu TT, Wang YY, Shu HB. The E3 ubiquitin ligase RNF5 targets virus-induced signaling adaptor for ubiquitination and degradation. J Immunol. 2010;184(11):6249-6255.
doi pubmed - Zhang ZD, Xiong TC, Yao SQ, Wei MC, Chen M, Lin D, Zhong B. RNF115 plays dual roles in innate antiviral responses by catalyzing distinct ubiquitination of MAVS and MITA. Nat Commun. 2020;11(1):5536.
doi pubmed - Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci U S A. 2007;104(18):7500-7505.
doi pubmed - Pan Y, Li R, Meng JL, Mao HT, Zhang Y, Zhang J. Smurf2 negatively modulates RIG-I-dependent antiviral response by targeting VISA/MAVS for ubiquitination and degradation. J Immunol. 2014;192(10):4758-4764.
doi pubmed - Moldogazieva NT, Lutsenko SV, Terentiev AA. Reactive oxygen and nitrogen species-induced protein modifications: implication in carcinogenesis and anticancer therapy. Cancer Res. 2018;78(21):6040-6047.
doi pubmed - Kim SY, Kim HJ, Kim HJ, Kim CH. Non-thermal plasma induces antileukemic effect through mTOR ubiquitination. Cells. 2020;9(3).
doi pubmed - Kim SY, Kim HJ, Kang SU, Kim YE, Park JK, Shin YS, Kim YS, et al. Non-thermal plasma induces AKT degradation through turn-on the MUL1 E3 ligase in head and neck cancer. Oncotarget. 2015;6(32):33382-33396.
doi pubmed - Wang Y, Zhao M, He S, Luo Y, Zhao Y, Cheng J, Gong Y, et al. Necroptosis regulates tumor repopulation after radiotherapy via RIP1/RIP3/MLKL/JNK/IL8 pathway. J Exp Clin Cancer Res. 2019;38(1):461.
doi pubmed - Scialo F, Fernandez-Ayala DJ, Sanz A. Role of mitochondrial reverse electron transport in ROS signaling: potential roles in health and disease. Front Physiol. 2017;8:428.
doi pubmed - Robb EL, Hall AR, Prime TA, Eaton S, Szibor M, Viscomi C, James AM, et al. Control of mitochondrial superoxide production by reverse electron transport at complex I. J Biol Chem. 2018;293(25):9869-9879.
doi pubmed - Yun JH, Yang YH, Han CH, Kang SU, Kim CH. Non-thermal atmospheric pressure plasma induces selective cancer cell apoptosis by modulating redox homeostasis. Cell Commun Signal. 2024;22(1):452.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.