| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://wjon.elmerpub.com |
Original Article
Volume 17, Number 2, April 2026, pages 247-255

Multi-Omics and Single-Cell Mendelian Randomization Reveal a Potential Role of VNN2 in Lung Adenocarcinoma in Resting Natural Killer Cells
Zhi Chun Xuea, b, Jing Linc, Jian Hui Wua, b, Zhi Wen Penga, b, Mei Yan Tanga, b, Peng Lianga, b, Hui Ling Chend, Gui Ju Fanga, b, e, Qing Xuea, b, e
aDepartment of Respiratory and Critical Care Medicine, Ningde Municipal Hospital of
Ningde Normal University, Ningde, Fujian, China
bDepartment of Respiratory and
Critical Care Medicine, Ningde Clinical Medical College of Fujian Medical University, Ningde,
Fujian, China
cDepartment of Emergency Medicine, Ningde Municipal Hospital of
Ningde Normal University, Ningde, Fujian, China
dCollege of Marine Sciences,
Ningde Normal University, Ningde, Fujian, China
eCorresponding Authors: Qing Xue
and Gui Ju Fang, Department of Respiratory and Critical Care Medicine, Ningde Municipal Hospital
of Ningde Normal University, Ningde, Fujian, China
Manuscript submitted October 4, 2025, accepted November 10, 2025, published online March 5,
2026
Short title: VNN2 and Resting NK Cells in Lung Adenocarcinoma
doi:
https://doi.org/10.14740/wjon2689
| Abstract | ▴Top |
Background: We aimed to evaluate the potential association between genetically predicted vanin-2 (VNN2) expression and lung adenocarcinoma (LUAD) risk, and to explore the immune cell subtype that may underlie this relationship.
Methods: We integrated whole-blood expression quantitative trait loci (eQTL) data from eQTLGen, plasma protein quantitative trait loci (pQTL) data from deCODE, and LUAD genome-wide association study (GWAS) data from European-ancestry cohorts, together with differential expression analysis using GEPIA2, to identify candidate genes for subsequent single-cell eQTL (sc-eQTL) Mendelian randomization (MR) analysis. For the sc-eQTL analysis, VNN2-associated eQTLs from 14 immune cell types profiled in the OneK1K single-cell eQTL resource were tested for associations with LUAD risk.
Results: Bulk-level MR analysis showed that genetically predicted increases in VNN2 expression and protein levels were significantly associated with a reduced risk of LUAD (eQTL-MR: odds ratio (OR) = 0.964, 95% confidence interval (95% CI), 0.934–0.995; P = 0.024; pQTL-MR: OR = 0.946, 95% CI, 0.921–0.970; P = 2.87 × 10−5). Transcriptomic analyses confirmed significant downregulation of VNN2 in LUAD tumors compared with normal lung tissues. sc-eQTL MR identified the strongest association in resting natural killer (rNK) cells (OR = 0.896, 95% CI, 0.829–0.967; P = 0.005).
Conclusions: Multi-omics and sc-eQTL MR analyses indicated that genetically predicted increases in VNN2 expression were associated with a reduced risk of LUAD, with the most pronounced effect observed in rNK cells. These findings suggest a potential cell type–specific role of VNN2 in LUAD susceptibility and warrant further studies to validate its biological relevance and clinical implications.
Keywords: VNN2; Lung adenocarcinoma; Mendelian randomization; sc-eQTL; Resting NK cells
| Introduction | ▴Top |
Lung adenocarcinoma (LUAD), the most common histologic subtype of lung cancer, remains a leading cause of cancer-related mortality worldwide [1, 2]. Despite diagnostic and therapeutic advances, the global incidence of LUAD continues to increase, and the 5-year survival rate remains low, especially among patients diagnosed at an advanced stage [3]. These observations underscore the urgent need to elucidate the molecular mechanisms underlying LUAD and to identify novel biomarkers for early detection and personalized therapy [4].
The tumor microenvironment (TME) plays a pivotal role in cancer progression, with immune cells, particularly natural killer (NK) cells, acting as key mediators of tumor surveillance and immune defense [5]. Among these subsets, resting natural killer (rNK) cells have gained increasing attention due to their roles in maintaining immune homeostasis and mediating early antitumor responses. However, the molecular mechanisms that regulate their function in LUAD remain poorly understood [6].
Vanin-2 (VNN2) encodes a glycosylphosphatidylinositol (GPI)-anchored membrane protein involved in inflammation, immune regulation, and hematopoietic functions [7, 8]. Although VNN2 has been proposed as a biomarker for immune-related and metabolic disorders, its role within the TME remains largely unexplored [9]. Most transcriptomic and expression quantitative trait locus (eQTL) studies of the TME have relied on bulk-level analyses, which lack the resolution to distinguish cell type–specific regulatory effects and may therefore overlook the contributions of immune cell heterogeneity to tumor progression [10, 11]. Recent advances in single-cell RNA sequencing (scRNA-seq) and single-cell eQTL (sc-eQTL) mapping offer powerful means to dissect cell type–specific regulatory mechanisms [12]. However, no direct evidence has linked the regulation of VNN2 expression within rNK cells to LUAD risk.
Recent sc-eQTL studies utilizing large-scale single-cell transcriptomic datasets have uncovered cell type–specific regulatory effects that are often overlooked in bulk analyses [13, 14]. In parallel, MR which integrates multi-omics data sources such as eQTL, pQTL, and genome-wide association study (GWAS) data, provides a powerful framework for inferring potential causal links between gene expression and disease risk [15]. Integrating sc-eQTL data with MR and colocalization analyses enables systematic investigation of cell type–specific genetic mechanisms at single-cell resolution [16, 17]. However, this integrative approach has not yet been applied to evaluate whether VNN2 expression within rNK cells is associated with the risk of LUAD.
In this study, we integrated sc-eQTL and Mendelian randomization (MR) analyses to evaluate whether genetically predicted VNN2 expression is associated with LUAD risk among different immune cell subtypes. This approach enabled us to explore the cell type–specific roles of VNN2 in immune regulation within the TME and to evaluate its potential relevance as a biomarker or therapeutic target. Given the emerging evidence of rNK cell involvement in LUAD, we speculated that any association would be most evident in rNK cells.
| Materials and Methods | ▴Top |
Study design
To clarify the analytical logic, we adopted a two-stage integrative MR framework to investigate the genetic determinants of LUAD using multi-omics data (Fig. 1). Stage 1 (bulk-level discovery): Whole-blood eQTL data from the eQTLGen Consortium and plasma protein pQTL data from the deCODE Genetics project were used as exposure datasets, while LUAD GWAS summary statistics (accession GCST004744) served as the outcome dataset. Multi-omics MR analyses were performed to identify genes whose genetically proxied expression or protein levels were associated with LUAD risk. To verify the transcriptomic direction and biological plausibility of these associations, GEPIA2—which integrates TCGA and GTEx transcriptomic data—was used to compare gene expression between LUAD tumor and normal tissues and to assess their prognostic relevance. Stage 2 (single-cell resolution): Genes identified from stage 1 were further evaluated using single-cell eQTL (sc-eQTL) data from the OneK1K project, which profiles genetic effects on gene expression across 14 peripheral immune cell types. Cell type–specific MR analyses were conducted to localize the bulk-level associations to specific immune subsets and to clarify potential immune-cell–mediated regulatory mechanisms. MR analyses were conducted under three core assumptions [18]: genetic instruments were strongly associated with the exposure; instruments were independent of both known and unknown confounders; and instruments influenced the outcome exclusively through the exposure, without horizontal pleiotropy.
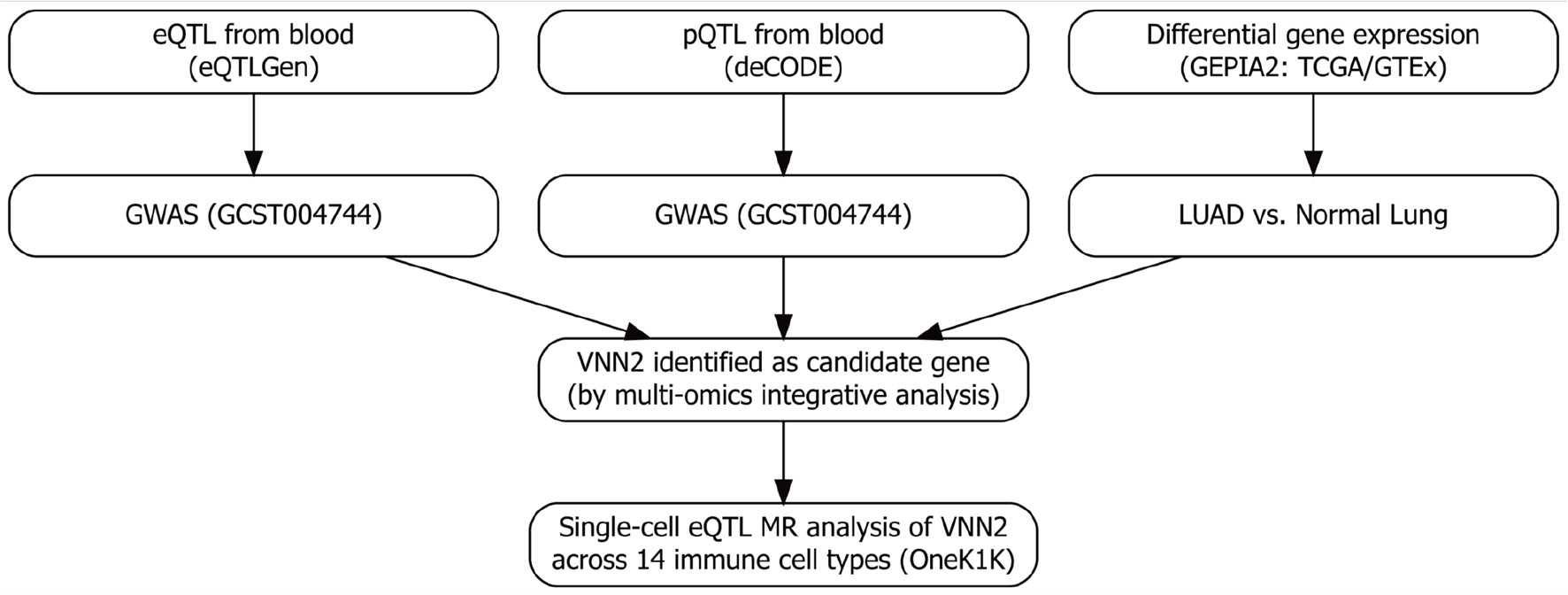 Click for large image |
Figure 1. Overview of the multi-omics Mendelian randomization (MR) framework. Stage 1: genome-wide screening integrating whole-blood eQTL (eQTLGen), plasma pQTL (deCODE), LUAD GWAS, and transcriptomic differential expression analyses (TCGA and GTEx via GEPIA2) to prioritize candidate genes. Stage 2: targeted single-cell MR analyses using the OneK1K sc-eQTL resource to evaluate cell type–specific associations with lung adenocarcinoma (LUAD) risk, with a focus on resting NK cells. eQTL: expression quantitative trait loci; NK: natural killer. |
Data sources and ethical statement
Five complementary public resources were integrated to support the two-stage MR framework: eQTLGen [19] provided genome-wide cis-eQTL summary data for whole blood and served as the transcriptomic exposure layer [20]. deCODE Genetics [21] offered plasma pQTL summary data, representing the proteomic exposure layer [22]. GWAS Catalog (LUAD accession GCST004744 [23]) provided the disease outcome data (11,273 LUAD cases and 55,483 controls; total n = 66,756) [24]. GEPIA2 [25] integrated TCGA and GTEx expression data to validate differential expression and survival associations of candidate genes. OneK1K project [26] offered single-cell eQTL data from 1.27 million peripheral blood mononuclear cells across 14 immune cell types, enabling cell type–specific MR analyses. Collectively, eQTLGen and deCODE were used to detect causal signals at the bulk level, GWAS data provided the LUAD outcome, GEPIA2 confirmed expression directionality and clinical relevance, and OneK1K refined these associations to specific immune subsets. All datasets were publicly available and de-identified, with ethics approval granted by the respective institutional review boards of the original studies. A detailed summary of dataset characteristics and analytical roles is provided in Supplementary Material 1 (wjon.elmerpub.com).
Instrument selection and quality control
Single nucleotide polymorphisms (SNPs) that met the genome-wide significance threshold (P < 5 × 10−8) were selected as instrumental variables (IVs) using a clumping window of 10,000 kb and a linkage disequilibrium (LD) threshold of r2 < 0.1. To minimize weak instrument bias, only SNPs with an F-statistic > 10 were retained. Palindromic and ambiguous SNPs were excluded from the analysis. All IVs were rigorously screened, which included removing SNPs associated with known confounders or outcome traits identified using the LDtrait tool [27] and the GWAS Catalog [28].
Differential expression analysis
Transcriptomic data from LUAD tumor and matched normal lung tissues were analyzed via the GEPIA2 platform. Differentially expressed genes were identified based on an absolute log2 fold change (|log2FC|) > 1 and a false discovery rate (FDR)-adjusted P < 0.05 [29].
Candidate gene prioritization
Candidate genes were prioritized through the integration of multi-omics evidence, including differential expression profiles, eQTL/pQTL associations, and MR estimates, emphasizing genes consistently supported across multiple analytical layers. Genes whose genetically predicted higher expression was associated with increased LUAD risk and that were upregulated in tumors were classified as putative oncogenes, whereas genes showing the opposite direction of association were considered putative tumor suppressors. This approach highlights candidates showing concordant directions between MR estimates and transcriptomic expression patterns, thereby prioritizing biologically meaningful targets.
Extraction and comparative analysis of sc-eQTLs
Following the identification of candidate genes through multi-omics genome-wide screening, we systematically characterized their genetic regulatory architecture across immune cell subpopulations. For each candidate gene, eQTL signals were retrieved from sc-eQTL summary statistics spanning 14 distinct immune cell types. The summary statistics for each cell type were iteratively processed to extract all SNP–gene pairs associated with the candidate genes. SNPs with nominal eQTL P < 0.05 were retained to capture potential regulatory variants. To ensure variant independence, linkage disequilibrium (LD) pruning (r2 < 0.1) was applied to remove highly correlated SNPs, resulting in the final set of independent eQTLs for each immune cell type.
MR analysis
Two-sample MR analyses were performed using the TwoSampleMR R package [18]. After rigorous quality control, bulk eQTL, pQTL, and sc-eQTL datasets were used as exposure datasets, with LUAD GWAS summary statistics as the outcome dataset. Causal estimates were obtained using the Wald ratio method for single-SNP instruments, whereas the inverse-variance weighted (IVW) approach was applied as the primary method for exposures with multiple SNP instruments. To account for potential horizontal pleiotropy, several robust MR methods were additionally implemented, including MR-Egger regression, the weighted median, simple mode, and weighted mode, to evaluate the robustness of causal estimates under different model assumptions. Horizontal pleiotropy was formally evaluated using the MR-Egger intercept test, whereas heterogeneity was assessed using Cochran’s Q statistic. Leave-one-out (LOO) analyses were further performed within the IVW framework. In each iteration, one SNP was removed from the instrument set to examine whether the overall causal estimate was disproportionately influenced by any single variant.
Transcriptomic and single-cell validation
Differential expression and survival analyses of VNN2 were conducted via the GEPIA2 platform [25], integrating TCGA and GTEx datasets. The Expression and Stage Plot modules were used to analyze differential expression and stage-specific variation of VNN2 across LUAD pathological stages (I–IV). The Survival Analysis module was used to assess the prognostic relevance of VNN2 expression based on Kaplan–Meier survival curves. To further investigate the cellular distribution of VNN2 within the TME, scRNA-seq data from the NSCLC dataset (GSE148071) were obtained from the TISCH2 database [30]. Violin plots generated within TISCH2 were used to depict cell type–specific expression levels across major immune and stromal cell populations.
Ethical statements
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. This study was conducted using publicly available summary-level data from genome-wide association studies (GWAS) and expression quantitative trait loci (eQTL), protein quantitative trait loci (pQTL), and single-cell eQTL (sc-eQTL) databases. All data sources used in this analysis obtained ethical approval from their respective institutional review boards, and no individual-level data were used.
| Results | ▴Top |
Genome-wide MR analysis identifies candidate genes associated with LUAD risk
To systematically evaluate the genetic determinants of LUAD, we integrated whole-blood eQTL, plasma pQTL, and European-ancestry LUAD GWAS summary statistics to perform genome-wide MR analyses. In the eQTL-based MR analysis, 1,798 genes showed nominally significant associations with LUAD risk (P < 0.05). Among these, 911 (50.7%) had positive β coefficients, indicating increased risk, whereas 887 (49.3%) had negative β coefficients, indicating reduced risk (Fig. 2a and Supplementary Material 2, wjon.elmerpub.com). In the pQTL-based MR analysis, 241 proteins were associated with LUAD risk at P < 0.05, including 127 (52.7%) with positive β coefficients and 114 (47.3%) with negative β coefficients (Fig. 2b and Supplementary Material 3, wjon.elmerpub.com).
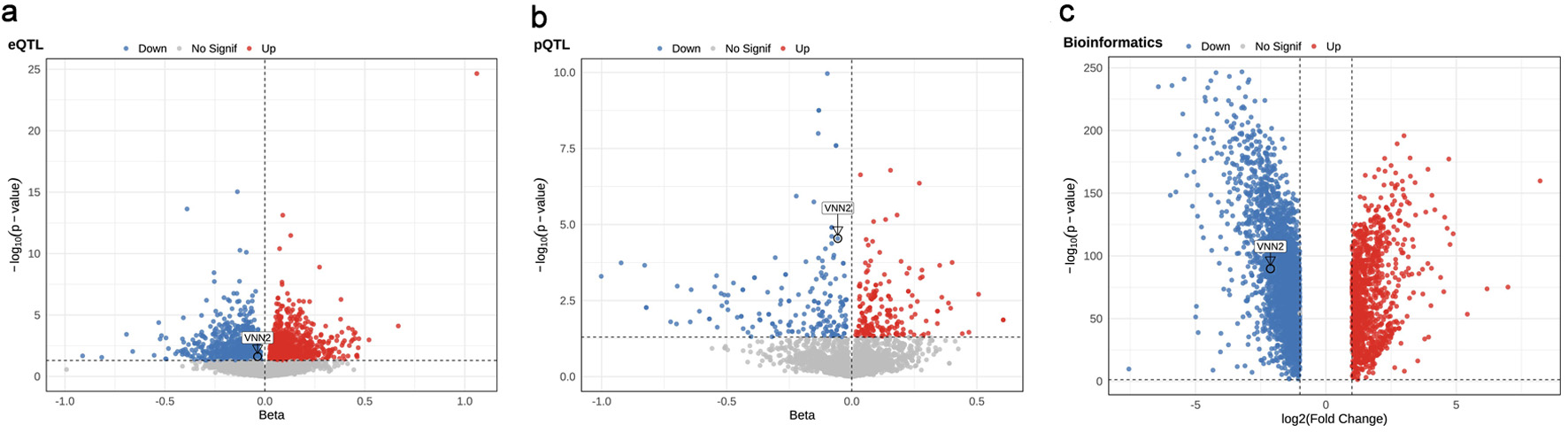 Click for large image |
Figure 2. Multi-omics identification of candidate genes associated with lung adenocarcinoma (LUAD). (a) eQTL-MR, (b) pQTL-MR, and (c) transcriptomic differential expression volcano plots highlight genes associated with LUAD risk. |
Differential expression in LUAD tissues
Differential gene expression analysis was performed using the GEPIA2 platform, which integrates transcriptomic data from The Cancer Genome Atlas (TCGA) and the Genotype-Tissue Expression (GTEx) project. A total of 4,245 genes were differentially expressed (|log2FC| > 1, FDR < 0.05), including 1,109 upregulated and 3,136 downregulated genes (Fig. 2c and Supplementary Material 3, wjon.elmerpub.com). Transcriptomic analysis revealed markedly reduced VNN2 mRNA expression in LUAD tumors relative to adjacent normal lung tissues (log2FC = –2.13, P = 1.55 × 10−90; Supplementary Material 4, wjon.elmerpub.com).
Cross-omics integration of candidate genes
To identify genes supported by convergent multi-omics evidence, we integrated results from eQTL-based MR, pQTL-based MR, and transcriptomic differential expression analyses. VNN2 showed consistent evidence across the three analyses. Transcriptomic analysis confirmed markedly reduced VNN2 mRNA expression in LUAD tumors relative to adjacent normal lung tissues (log2FC = –2.13, P = 1.55 × 10−90; Supplementary Material 5, wjon.elmerpub.com). In eQTL-based MR analysis, genetically predicted higher VNN2 expression was significantly associated with reduced LUAD risk (β = –0.036, OR = 0.964, 95% CI: 0.934–0.995, P = 0.024; Supplementary Material 2, wjon.elmerpub.com). Consistent findings were obtained in pQTL-based MR analysis, in which genetically predicted higher VNN2 protein levels were significantly associated with reduced LUAD risk (β = –0.056, OR = 0.946, 95% CI: 0.921–0.970, P = 2.87 × 10−5; Supplementary Material 3, wjon.elmerpub.com).
Cell type–specific MR analysis of VNN2 in peripheral immune cell subsets
To investigate whether the genetic association between VNN2 expression and LUAD risk exhibits cell type–specific effects, we performed single-cell eQTL–based MR analysis across 14 peripheral blood immune cell types. In the OneK1K dataset, cis-eQTLs for VNN2 meeting genome-wide significance and instrument strength thresholds were identified in only six immune cell types—CD4+SOX4+ T, CD8+ effector T, classical monocytes, nonclassical monocytes, NK cells, and rNK cells. In the remaining eight immune cell types, no significant VNN2-associated SNPs were detected, indicating the absence of valid genetic instruments for MR analysis. For each cell type, only independent eQTLs (P < 0.05 after LD clumping) were retained as instrumental variables. Genetically predicted higher VNN2 expression in rNK cells was significantly associated with reduced LUAD risk (OR = 0.75, 95% CI: 0.57–0.98, P = 0.039). No statistically significant associations were observed in other immune cell types (Fig. 3 and Supplementary Material 5, wjon.elmerpub.com).
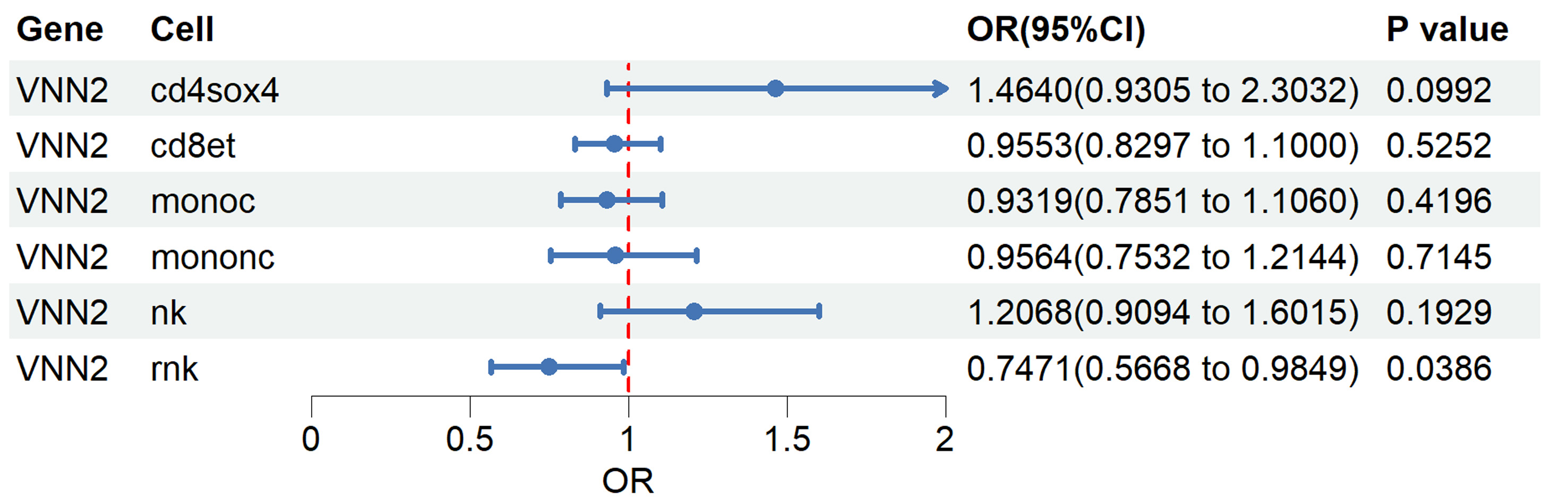 Click for large image |
Figure 3. Single-cell Mendelian randomization (MR) analysis of VNN2 across immune cell types. Overview of the OneK1K dataset comprising 14 peripheral blood immune cell types profiled for eQTLs. Forest plot showing MR estimates of genetically predicted VNN2 expression and lung adenocarcinoma (LUAD) risk across immune cell subsets. eQTL: expression quantitative trait loci. |
Sensitivity analyses
To evaluate the robustness of the MR findings, we performed a series of sensitivity analyses. Using robust estimators such as MR-Egger regression, weighted median, and weighted mode, the causal estimates for VNN2 expression and LUAD risk were directionally consistent with those obtained from the IVW analysis, supporting the robustness of the causal inference (Supplementary Material 6, wjon.elmerpub.com). The MR-Egger intercept test showed no evidence of directional pleiotropy, and Cochran’s Q statistic indicated no substantial heterogeneity among the SNP instruments. Moreover, LOO analyses showed that excluding any single SNP did not disproportionately alter the IVW estimates for either eQTL- or pQTL-based analyses (Supplementary Material 7, wjon.elmerpub.com), confirming that no single variant drove the observed associations.
We further evaluated the expression pattern and clinical relevance of VNN2 using publicly available transcriptomic and single-cell datasets. Kaplan–Meier survival analysis using the GEPIA2 database showed no significant difference in overall survival between patients with high and low VNN2 expression (log-rank P = 0.66, hazard ratio (HR) = 0.94; Supplementary Material 8, wjon.elmerpub.com). Stage-specific analysis using the GEPIA2 Stage Plot module indicated no significant differences in VNN2 expression among pathological stages I–IV of LUAD (analysis of variance (ANOVA) P = 0.964; Supplementary Material 9, wjon.elmerpub.com). To visualize the cellular distribution of VNN2 within the TME, we examined the non-small cell lung cancer (NSCLC) scRNA-seq dataset (GSE148071) available in the TISCH2 database. As shown in Supplementary Material 10 (wjon.elmerpub.com), VNN2 expression was relatively higher in immune cell populations—particularly monocytes and macrophages—while being nearly absent in epithelial and malignant cells. Notably, although rNK cells were not individually annotated in this dataset, the overall immune cell–restricted expression pattern of VNN2 was consistent with its proposed immunoregulatory role in the TME.
| Discussion | ▴Top |
In this study, we systematically integrated multi-omics MR analyses with sc-eQTL data to investigate genetic associations linking VNN2 expression with LUAD risk. Our analyses showed that higher genetically predicted VNN2 expression was significantly associated with reduced LUAD risk at both transcriptomic and proteomic levels, consistent with its downregulation in LUAD tumor tissues relative to normal lung tissues. The detection of VNN2 in plasma likely reflects its soluble, cleaved form released from leukocyte membranes, as previously reported for phospholipase-mediated GPI-anchor hydrolysis. Furthermore, single-cell MR analysis showed that this association was most pronounced in rNK cells, suggesting a potential cell type–specific involvement in tumor immunity. These findings extend previous bulk-level omics observations to single-cell resolution and provide exploratory genetic evidence that may inform future mechanistic studies and NK cell–targeted therapeutic strategies.
Bulk transcriptomic analysis (TCGA/GTEx via GEPIA2) indicated lower VNN2 expression in LUAD tumor tissues relative to normal lung tissues; however, this pattern should not be interpreted as indicating that tumor cells are the principal source of VNN2. VNN2 (Vanin-2, GPI-80) is a GPI-anchored membrane protein primarily expressed on leukocytes—particularly neutrophils and monocytes—where it regulates cell adhesion and migration [31, 32]. ScRNA-seq data from the TISCH2 NSCLC dataset (Supplementary Material 10, wjon.elmerpub.com) showed that VNN2 expression was largely restricted to immune cell populations—especially monocytes and macrophages—while epithelial and malignant cells displayed minimal or nearly absent expression. Therefore, the reduced VNN2 signal in bulk LUAD samples likely reflects decreased immune-cell infiltration or immunosuppressive remodeling of the TME, rather than tumor-intrinsic downregulation. Previous studies have mainly examined the role of VNN2 in hematological disorders; for example, high VNN2 expression has been associated with chemoresistance in pediatric B-cell acute lymphoblastic leukemia [33]. In solid tumors, limited evidence links VNN2 to cancer progression—for instance, it has been included as a prognostic factor in hepatocellular carcinoma models, shown to regulate tumor invasion in osteosarcoma via miR-106a targeting, and associated with poor prognosis in metastatic renal cancer through high expression in peripheral myeloid cells [34–36]. Preliminary evidence also suggests that VNN2 may influence tumor redox status and inflammatory pathways through its enzymatic activity, but mechanistic studies are scarce [37], particularly in the context of LUAD and the tumor immune microenvironment. Beyond tumor-intrinsic roles, recent studies have highlighted potential immunological functions of VNN2. Notably, its high expression in monocytic myeloid-derived suppressor cells (Mo-MDSCs) suggests a possible involvement in immunosuppressive states [38]. To date, however, no direct evidence has demonstrated that VNN2 regulates NK-cell function. NK cells are central to immune surveillance in lung cancer, and their tumor infiltration correlates closely with patient prognosis [39]. While NK-cell dysfunction is a recognized mechanism of tumor immune evasion, the relationship between VNN2, NK cells, and LUAD risk has yet to be systematically examined. In this study, we integrated MR with sc-eQTL analysis to investigate whether genetically predicted VNN2 expression was associated with LUAD risk across immune cell subtypes. This association was most pronounced in rNK cells, consistent with a potential cell type–specific role of VNN2 in tumor immunity. By extending bulk-level omics analyses of lung cancer to single-cell resolution [40, 41], this work provides genetic evidence for a potential immunoregulatory role of VNN2 that may inform future mechanistic studies on NK-cell–related immune pathways.
Together with previous evidence, our findings suggest that higher VNN2 expression may be linked to a lower risk of LUAD, potentially by modulating the functional state of rNK cells and contributing to immune surveillance. VNN2 encodes GPI-80, a GPI-anchored protein with pantetheinase activity, which has been implicated in the regulation of oxidative stress and inflammatory pathways, possibly through activation of the NF-κB and IL-1β pathways [42, 43]. Elevated VNN2 expression has been associated with increased IL-1β levels and sustained NF-κB activation [44]. These pro-inflammatory mediators (e.g., IL-1β, type I interferons) could “prime” rNK cells [45], enhancing the expression of activating receptors (e.g., NKG2D, NKp44) and strengthening their capacity for tumor recognition. Resting NK cells serve as a reservoir for activated NK cells, and their abundance and activation capacity are critical for maintaining immune surveillance; however, excessive activation may indicate immune exhaustion. Epidemiological and experimental studies have linked higher NK-cell activity or tumor infiltration to reduced cancer risk and improved prognosis [46]. In addition, VNN2 may influence NK-cell function indirectly by modulating immune-cell migration, adhesion, and metabolic state. For example, VNN2 has been reported to regulate leukocyte β2-integrin (CD11b/CD18)–mediated adhesion and migration [47, 48], potentially facilitating NK-cell recruitment and retention within tumor tissues. Its pantetheinase activity generates cysteamine and pantothenic acid [45], which can modulate glutathione and reactive oxygen species (ROS) levels, possibly supporting NK-cell survival and activity under oxidative stress. While these mechanisms provide biologically plausible explanations for the observed cell type–specific genetic association, experimental validation—such as NK-cell co-culture assays and in vivo models—will be necessary to confirm their relevance.
This study has several limitations that should be acknowledged. First, although extensive sensitivity analyses were conducted, the MR results may still be susceptible to residual pleiotropy. Second, the genetic instruments were primarily derived from European-ancestry eQTL and pQTL datasets, which may limit the generalizability of our findings to other ancestries and lung tissue–specific regulatory landscapes. Third, compared with bulk eQTL analyses, single-cell eQTL–based MR was limited by smaller sample sizes, reduced statistical power, and potential biases in cell type annotation. Finally, although the association between VNN2 and LUAD risk was most pronounced in rNK cells, the underlying biological mechanisms require experimental validation. Additionally, survival stratification according to NK cell–specific VNN2 expression could not be evaluated because the available single-cell datasets lack patient follow-up information.
Furthermore, transcriptomic validation using the GEPIA2 database revealed no significant difference in overall survival between patients with high and low VNN2 expression (log-rank P = 0.66; Supplementary Material 8, wjon.elmerpub.com), indicating that the protective genetic effect of VNN2 on LUAD risk may not be directly reflected at the transcriptional level. In addition, single-cell transcriptomic analysis from the TISCH2 database showed that VNN2 expression was predominantly enriched in immune cell populations—particularly monocytes and macrophages—whereas epithelial and malignant cells exhibited minimal expression (Supplementary Material 10, wjon.elmerpub.com). Although rNK cells were not individually annotated in this dataset, the immune cell–restricted expression pattern of VNN2 is consistent with our sc-eQTL MR findings and supports its potential role in immune regulation within the LUAD microenvironment.
In addition, clinical factors such as tumor stage, grade, and treatment history were not adjusted in this study because the MR analyses were performed using publicly available summary-level data that lack individual-level clinical annotations. Nevertheless, the MR framework inherently mitigates potential confounding from these factors, as germline genetic variants are randomly allocated at conception and are independent of disease progression. Supporting evidence from the GEPIA2 stage analysis showed that VNN2 expression levels did not significantly differ across LUAD stages (stage I–IV; ANOVA P = 0.964), suggesting that tumor stage heterogeneity is unlikely to bias our findings. Future studies integrating individual-level clinical data or implementing multivariable Mendelian randomization (MVMR) frameworks are warranted to further validate the causal relationship between VNN2 expression and LUAD risk and to clarify its translational relevance in the clinical setting. This study focused on the genetic association between VNN2 expression and LUAD susceptibility rather than its prognostic or predictive value. Future clinical and longitudinal studies are warranted to determine whether VNN2 expression may serve as a biomarker for patient outcomes or therapeutic response.
Conclusions
Multi-omics analyses indicated that genetically predicted higher VNN2 expression was associated with a reduced risk of LUAD, most notably in rNK cells. These results highlight a potential cell type–specific role of VNN2 in LUAD susceptibility and immune regulation. The findings should be regarded as hypothesis-generating, and further functional and clinical studies are needed to validate the biological mechanisms and assess any potential clinical relevance.
| Supplementary Material | ▴Top |
Suppl 1. Summary of data sources and their analytical roles in the two-stage integrative framework.
Suppl 2. Two-sample MR analysis of blood eQTLs associated with LUAD (P < 0.05).
Suppl 3. Two-sample MR analysis of blood pQTLs associated with LUAD (P < 0.05).
Suppl 4. Differentially expressed genes between LUAD tumor and normal tissues (GEPIA2).
Suppl 5. Two-sample MR estimates of the association between genetically predicted VNN2 expression and LUAD risk across 14 immune cell types.
Suppl 6. Summary of two-sample Mendelian randomization estimates for the association between genetically predicted VNN2 expression/protein levels and LUAD risk using eQTL, pQTL, and sc-eQTL instruments.
Suppl 7. Leave-one-out sensitivity analyses for the association between VNN2 and LUAD risk. (A) eQTL-based MR analysis. (B) pQTL-based MR analysis.
Suppl 8. Kaplan–Meier survival curve showing the association between VNN2 expression and overall survival in LUAD patients based on TCGA data (GEPIA2).
Suppl 9. Expression of VNN2 across pathological stages of lung adenocarcinoma (LUAD) based on TCGA data (GEPIA2 platform).
Suppl 10. Violin plot illustrating VNN2 expression across major immune and stromal cell populations in NSCLC (GSE148071, TISCH2).
Acknowledgments
We thank all contributors and collaborators for their valuable support and assistance during this study. We also gratefully acknowledge the eQTLGen Consortium, deCODE project, OneK1K resource, The Cancer Genome Atlas (TCGA), the Genotype-Tissue Expression (GTEx) project, the GWAS Catalog, and the GEPIA2 platform for providing access to valuable datasets that made this research possible.
Financial Disclosure
This work was supported by the Natural Science Foundation of Ningde (grant numbers 2023J10, 2024J19) and the Fujian Provincial Key Clinical Specialty Construction Project in 2022 (Fujian Medical Policy Letter (2022) No. 884).
Conflict of Interest
The authors of this work have nothing to disclose.
Informed Consent
No additional ethical approval or informed consent was required.
Author Contributions
Conception and design: Zhi Chun Xue, Gui Ju Fang, and Qing Xue; administrative support: Hui Ling Chen; provision of study materials or patients: not applicable; collection and assembly of data: Jing Lin, Jian Hui Wu, Zhi Wen Peng, Mei Yan Tang, Peng Liang; data analysis and interpretation: Zhi Chun Xue, Jing Lin, Jian Hui Wu, Zhi Wen Peng, Mei Yan Tang, and Peng Liang; manuscript writing: all authors; final approval of manuscript: all authors.
Data Availability
All datasets used in this study are publicly available, including eQTLGen (whole-blood eQTL), deCODE (plasma pQTL), LUAD GWAS (European ancestry cohorts), GEPIA2 integrating TCGA and GTEx (transcriptomics), and OneK1K (single-cell eQTL). The analysis code and additional materials are available from the corresponding authors upon request.
| References | ▴Top |
- Xin S, Wen M, Tian Y, Dong H, Wan Z, Jiang S, Meng F, et al. Impact
of histopathological subtypes on invasive lung adenocarcinoma: from epidemiology to tumour
microenvironment to therapeutic strategies. World J Surg Oncol.
2025;23(1):66.
doi pubmed - Dantas E, Murthy A, Ahmed T, Ahmed M, Ramsamooj S, Hurd MA, Lam T, et
al. TIMP1 is an early biomarker for detection and prognosis of lung cancer. Clin Transl Med.
2023;13(10):e1391.
doi pubmed - Willner J, Narula N, Moreira AL. Updates on lung adenocarcinoma:
invasive size, grading and STAS. Histopathology. 2024;84(1):6-17.
doi pubmed - Davies MPA, Sato T, Ashoor H, Hou L, Liloglou T, Yang R, Field JK.
Plasma protein biomarkers for early prediction of lung cancer. EBioMedicine.
2023;93:104686.
doi pubmed - Wang C, Yu Q, Song T, Wang Z, Song L, Yang Y, Shao J, et al. The
heterogeneous immune landscape between lung adenocarcinoma and squamous carcinoma revealed by
single-cell RNA sequencing. Signal Transduct Target Ther. 2022;7(1):289.
doi pubmed - Pelletier A, Nelius E, Fan Z, Khatchatourova E, Alvarado-Diaz A, He
J, Krzywinska E, et al. Resting natural killer cell homeostasis relies on tryptophan/NAD(+)
metabolism and HIF-1alpha. EMBO Rep. 2023;24(6):e56156.
doi pubmed - Bornhauser B, Cario G, Rinaldi A, Risch T, Rodriguez Martinez V,
Schutte M, Warnatz HJ, et al. The hematopoietic stem cell marker VNN2 is associated with
chemoresistance in pediatric B-cell precursor ALL. Blood Adv. 2020;4(17):4052-4064.
doi pubmed - Liu C, Alimu X, Zeng X, Bahabayi A, Gao Y, Hu Y, Chen Y, et al.
Vanin-2 is expressed in peripheral blood T cells and upregulated in patients with systemic lupus
erythematosus. J Leukoc Biol. 2024;116(6):1469-1478.
doi pubmed - Bahabayi A, Alimu X, Wang G, Gao Y, Chen Y, Zhao J, Lian X, et al.
VNN2-expressing circulating monocytes exhibit unique functional characteristics and are
decreased in patients with primary Sjogren's syndrome. J Autoimmun.
2024;147:103275.
doi pubmed - Kang B, Camps J, Fan B, Jiang H, Ibrahim MM, Hu X, Qin S, et al.
Parallel single-cell and bulk transcriptome analyses reveal key features of the gastric tumor
microenvironment. Genome Biol. 2022;23(1):265.
doi pubmed - Tan Z, Chen X, Zuo J, Fu S, Wang H, Wang J. Comprehensive analysis of
scRNA-Seq and bulk RNA-Seq reveals dynamic changes in the tumor immune microenvironment of
bladder cancer and establishes a prognostic model. J Transl Med. 2023;21(1):223.
doi pubmed - Ding S, Chen X, Shen K. Single-cell RNA sequencing in breast cancer:
Understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun
(Lond). 2020;40(8):329-344.
doi pubmed - Yazar S, Alquicira-Hernandez J, Wing K, Senabouth A, Gordon MG,
Andersen S, Lu Q, et al. Single-cell eQTL mapping identifies cell type-specific genetic control
of autoimmune disease. Science. 2022;376(6589):eabf3041.
doi pubmed - Chen C, Cai Y, Hu W, Tan K, Lu Z, Zhu X, Liu Z, et al. Single-cell
eQTL Mapping Reveals Cell Subtype-specific Genetic Control and Mechanism in Malignant
Transformation of Colorectal Cancer. Cancer Discov. 2025;15(8):1649-1675.
doi pubmed - Ray A, Alabarse P, Malik R, Sargurupremraj M, Bernhagen J, Dichgans
M, Baumeister SE, et al. Single-cell transcriptome-wide Mendelian randomization and
colocalization analyses uncover cell-specific mechanisms in atherosclerotic cardiovascular
disease. Am J Hum Genet. 2025;112(7):1597-1609.
doi pubmed - Ying H, Wu X, Jia X, Yang Q, Liu H, Zhao H, Chen Z, et al.
Single-cell transcriptome-wide Mendelian randomization and colocalization reveals
immune-mediated regulatory mechanisms and drug targets for COVID-19. EBioMedicine.
2025;113:105596.
doi pubmed - Hong Y, Wang Y, Shu W, Li J, Mi Y, Chen H, Chen C. Mapping the
immune-genetic architecture of aging: A single-cell causal framework for biomarker discovery and
therapeutic targeting. Ageing Res Rev. 2025;111:102835.
doi pubmed - Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian
randomization: using genes as instruments for making causal inferences in epidemiology. Stat
Med. 2008;27(8):1133-1163.
doi pubmed - https://www.eqtlgen.org/cis-eqtls.html.
- Võsa U, Claringbould A, Westra HJ, Bonder MJ, Deelen P, et al. Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv. 2018;19:447367.
- https://www.decode.com/summarydata/.
- Ferkingstad E, Sulem P, Atlason BA, Sveinbjornsson G, Magnusson MI,
Styrmisdottir EL, Gunnarsdottir K, et al. Large-scale integration of the plasma proteome with
genetics and disease. Nat Genet. 2021;53(12):1712-1721.
doi pubmed - https://www.ebi.ac.uk/gwas/studies/GCST004744.
- McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC,
Caporaso NE, et al. Large-scale association analysis identifies new lung cancer susceptibility
loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet.
2017;49(7):1126-1132.
doi pubmed - http://gepia2.cancer-pku.cn/.
- https://onek1k.org/.
- https://ldlink.nci.nih.gov/.
- Burgess S, Thompson SG. Interpreting findings from Mendelian
randomization using the MR-Egger method. Eur J Epidemiol.
2017;32(5):377-389.
doi pubmed - Wang X, Pei Z, Hao T, Ariben J, Li S, He W, Kong X, et al. Prognostic
analysis and validation of diagnostic marker genes in patients with osteoporosis. Front Immunol.
2022;13:987937.
doi pubmed - http://tisch.comp-genomics.org/.
- Yoshitake H, Takeda Y, Nitto T, Sendo F. Cross-linking of GPI-80, a
possible regulatory molecule of cell adhesion, induces up-regulation of CD11b/CD18 expression on
neutrophil surfaces and shedding of L-selectin. J Leukoc Biol.
2002;71(2):205-211.
pubmed - Kaskow BJ, Proffitt JM, Blangero J, Moses EK, Abraham LJ. Diverse
biological activities of the vascular non-inflammatory molecules - the Vanin pantetheinases.
Biochem Biophys Res Commun. 2012;417(2):653-658.
doi pubmed - Izzi V, Lakkala J, Devarajan R, Savolainen ER, Koistinen P,
Heljasvaara R, Pihlajaniemi T. Vanin 1 (VNN1) levels predict poor outcome in acute myeloid
leukemia. Am J Hematol. 2018;93(1):E4-E7.
doi pubmed - Li W, Lu J, Ma Z, Zhao J, Liu J. An integrated model based on a
six-gene signature predicts overall survival in patients with hepatocellular carcinoma. Front
Genet. 2019;10:1323.
doi pubmed - Chen Y, Huang T, Yang X, Liu C, Li P, Wang Z, Zhi S. MicroRNA-106a
regulates the proliferation and invasion of human osteosarcoma cells by targeting VNN2. Oncol
Rep. 2018;40(4):2251-2259.
doi pubmed - Kato T, Takeda Y, Ito H, Kurota Y, Yamagishi A, Sakurai T, Naito S,
et al. GPI-80 as a useful index for myeloid cell heterogeneity and a potential prognostic
biomarker for metastatic renal cell carcinoma. Tohoku J Exp Med.
2019;249(3):203-212.
doi pubmed - Takeda Y, Kurota Y, Kato T, Ito H, Araki A, Nara H, Saitoh S, et al.
GPI-80 augments NF-kappaB activation in tumor cells. Int J Mol Sci.
2021;22(21):12027.
doi pubmed - Soler DC, Kerstetter-Fogle A, Young AB, Rayman P, Finke JH, Debanne
SM, Cooper KD, et al. Healthy myeloid-derived suppressor cells express the surface ectoenzyme
Vanin-2 (VNN2). Mol Immunol. 2022;142:1-10.
doi pubmed - Szentkereszty M, Ladanyi A, Galffy G, Tovari J, Losonczy G. Density
of tumor-infiltrating NK and Treg cells is associated with 5 years progression-free and overall
survival in resected lung adenocarcinoma. Lung Cancer. 2024;192:107824.
doi pubmed - Zhang L, Xiong Y, Zhang J, Feng Y, Xu A. Systematic proteome-wide
Mendelian randomization using the human plasma proteome to identify therapeutic targets for lung
adenocarcinoma. J Transl Med. 2024;22(1):330.
doi pubmed - Ding Z, Chen J, Li B, Ji X. Inflammatory factors and risk of lung
adenocarcinoma: a Mendelian randomization study mediated by blood metabolites. Front Endocrinol
(Lausanne). 2024;15:1446863.
doi pubmed - Nitto T, Onodera K. Linkage between coenzyme a metabolism and
inflammation: roles of pantetheinase. J Pharmacol Sci. 2013;123(1):1-8.
doi pubmed - Bartucci R, Salvati A, Olinga P, Boersma YL. Vanin 1: its
physiological function and role in diseases. Int J Mol Sci.
2019;20(16):3891.
doi pubmed - Yu W, Hu S, Yang R, Lin L, Mao C, Jin M, Gu Y, et al. Upregulated
Vanins and their potential contribution to periodontitis. BMC Oral Health.
2022;22(1):614.
doi pubmed - Mattiola I, Pesant M, Tentorio PF, Molgora M, Marcenaro E, Lugli E,
Locati M, et al. Priming of Human Resting NK Cells by Autologous M1 Macrophages via the
Engagement of IL-1beta, IFN-beta, and IL-15 Pathways. J Immunol.
2015;195(6):2818-2828.
doi pubmed - Zhang S, Liu W, Hu B, Wang P, Lv X, Chen S, Shao Z. Prognostic
significance of tumor-infiltrating natural killer cells in solid tumors: a systematic review and
meta-analysis. Front Immunol. 2020;11:1242.
doi pubmed - Suzuki K, Watanabe T, Sakurai S, Ohtake K, Kinoshita T, Araki A,
Fujita T, et al. A novel glycosylphosphatidyl inositol-anchored protein on human leukocytes: a
possible role for regulation of neutrophil adherence and migration. J Immunol.
1999;162(7):4277-4284.
pubmed - Sendo D, Takeda Y, Ishikawa H, Sendo F, Araki Y. Localization of
GPI-80, a beta2-integrin-associated glycosylphosphatidyl-inositol anchored protein, on strongly
CD14-positive human monocytes. Immunobiology. 2003;207(3):217-221.
doi pubmed
This
article is distributed under the terms of the Creative Commons Attribution 4.0 International
License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any
medium, including commercial use, provided the original work is properly
cited.
World Journal of Oncology is published by Elmer Press Inc.