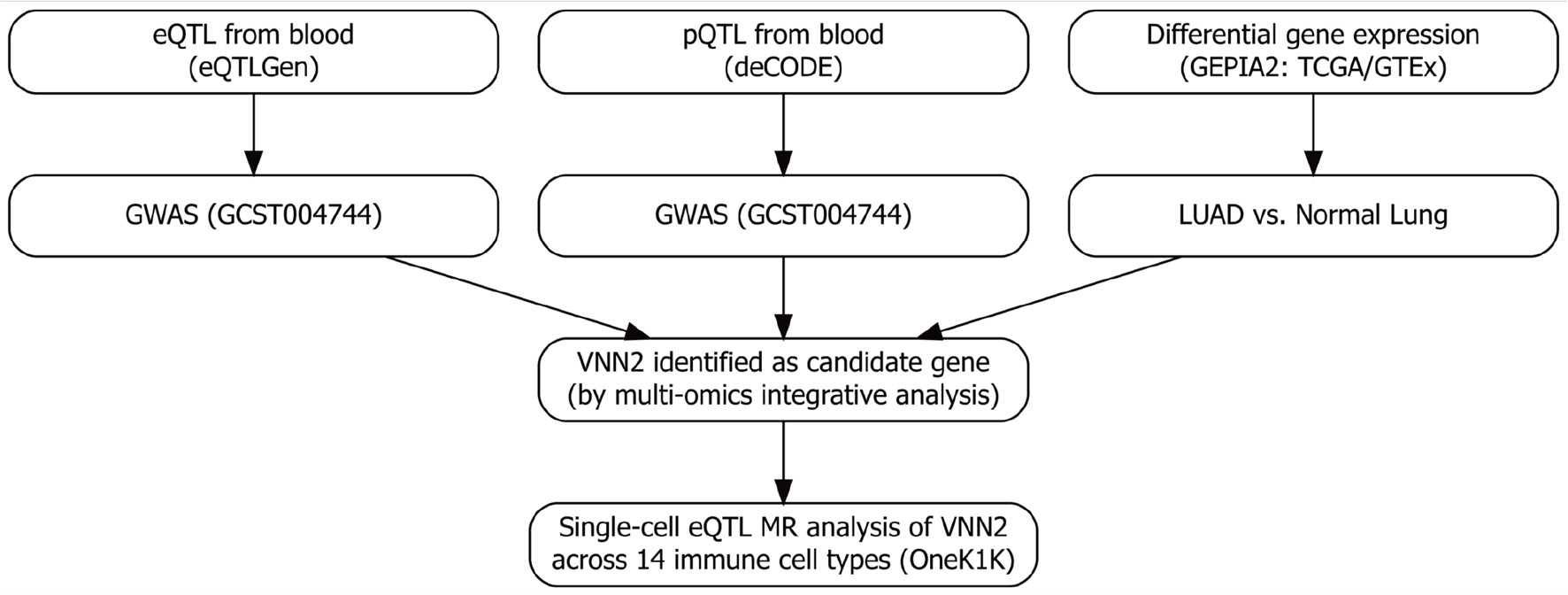
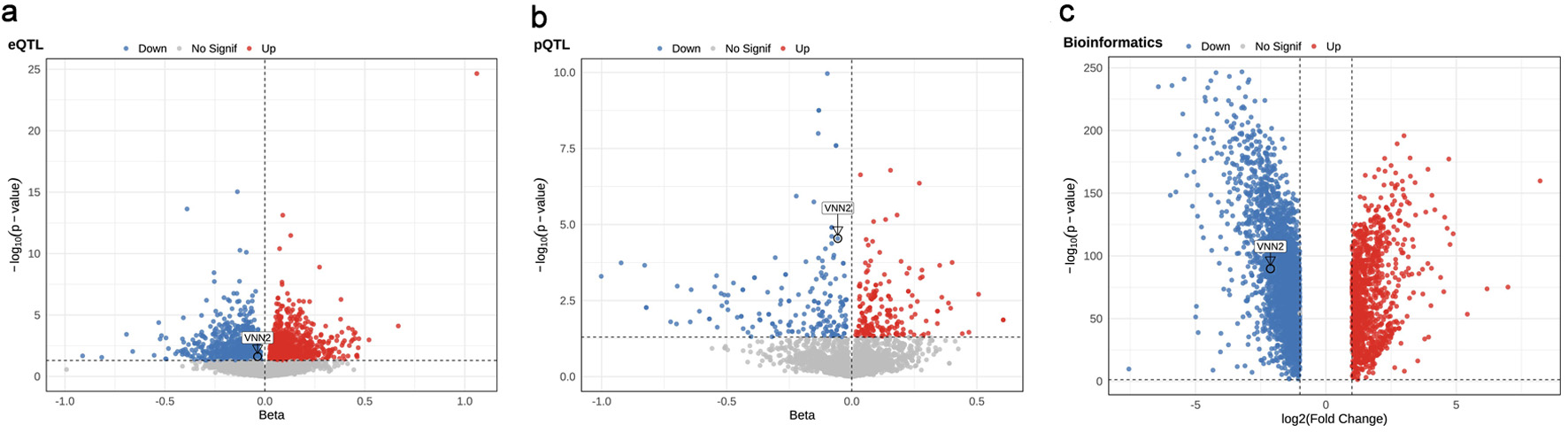
↓ Figure 1. Overview of the multi-omics Mendelian
randomization (MR) framework. Stage 1: genome-wide screening integrating whole-blood eQTL (eQTLGen),
plasma pQTL (deCODE), LUAD GWAS, and transcriptomic differential expression analyses (TCGA and GTEx via
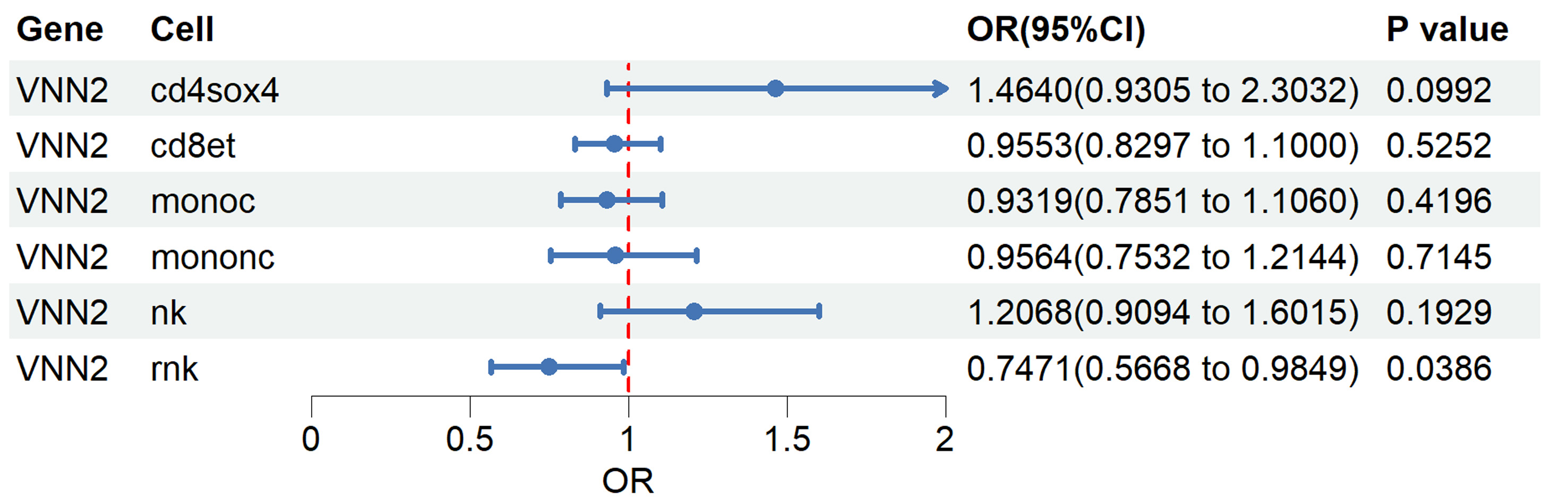
GEPIA2) to prioritize candidate genes. Stage 2: targeted single-cell MR analyses using the OneK1K
sc-eQTL resource to evaluate cell type–specific associations with lung adenocarcinoma (LUAD)
risk, with a focus on resting NK cells. eQTL: expression quantitative trait loci; NK: natural
killer.