| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://wjon.elmerpub.com |
Review
Volume 000, Number 000, September 2025, pages 000-000

IgM Myeloma: A Comprehensive Overview and Practical Approach to Chemotherapeutic Management
Hadeel Elwaheidia, c, Alaa Hamada, Farah Salameha, Fadwa Elkordya, Rojina FathAlrahmana, Amr Hanbalib
aCollege of Medicine, Alfaisal University, Riyadh, Saudi Arabia
bDepartment of Hematology, King Faisal Specialist Hospital & Research Center, Riyadh, Saudi Arabia
cCorresponding Author: Hadeel Elwaheidi, College of Medicine, Alfaisal University, Riyadh, Saudi Arabia
Manuscript submitted May 19, 2025, accepted August 13, 2025, published online September 3, 2025
Short title: IgM Myeloma: From Diagnosis to Chemotherapy
doi: https://doi.org/10.14740/wjon2611
- Abstract
- Introduction
- Methodology
- Pathophysiology
- Clinical Features
- Diagnostics
- Prognosis
- Therapeutic Management
- Supportive Management
- Conclusion
- References
| Abstract | ▴Top |
IgM myeloma is a rare subtype of multiple myeloma (MM) comprising 0.5% of all of its cases. It is characterized by the unregulated proliferation of IgM-secreting plasma cells in the bone marrow. The underlying pathogenesis involves dysregulation of isotype switch recombination, leading to various translocations involving chromosomes such as 11q13 and 4p16. Patients usually present with symptoms of hyperviscosity syndrome, bone marrow infiltration, and organomegaly. Diagnostic workup includes clinical evaluation, laboratory tests (electrophoresis, bone marrow biopsy, cytogenetics, immunohistochemistry), and imaging. Treatment options for IgM myeloma include proteasome inhibitors, immunomodulatory drugs, monoclonal antibodies, and autologous stem cell transplantation. However, no clear management guidelines are established for this rare subtype of MM. This article provides an up-to-date detailed overview of the pathogenesis, clinical features, and diagnostics of IgM myeloma.
Keywords: Multiple myeloma; IgM myeloma; Management; Chemotherapy; IgM
| Introduction | ▴Top |
Multiple myeloma (MM) is an entity characterized by the clonal and neoplastic proliferation of plasma cells in the bone marrow. It is considered by the World Health Organization as a lymphoproliferative B-cell disease [1]. MM accounts for 1% of cancers worldwide and 10% of all hematological cancers [2, 3]. In 2018, the global age-standardized incidence of MM was 2.1 per 100,000 [4]. MM evolves on the basis of a premalignant precursor termed “monoclonal gammopathy of undetermined significance” (MGUS). MGUS is diagnosed incidentally in 3-5% of people above the age of 50. It then proceeds to progress to MM at a rate of 1% per annum [5, 6]. MGUS can occasionally progress to a transitional phase named smoldering myeloma, which has a higher risk of further developing into the overt and symptomatic MM [7]. In the first 5 years of diagnosing smoldering myeloma, the risk of it developing into MM is 10% per year [8]. The diagnostic criteria of the International Myeloma Working Group (IMWG), for each of the three gammopathies, are summarized in Figure 1 [9].
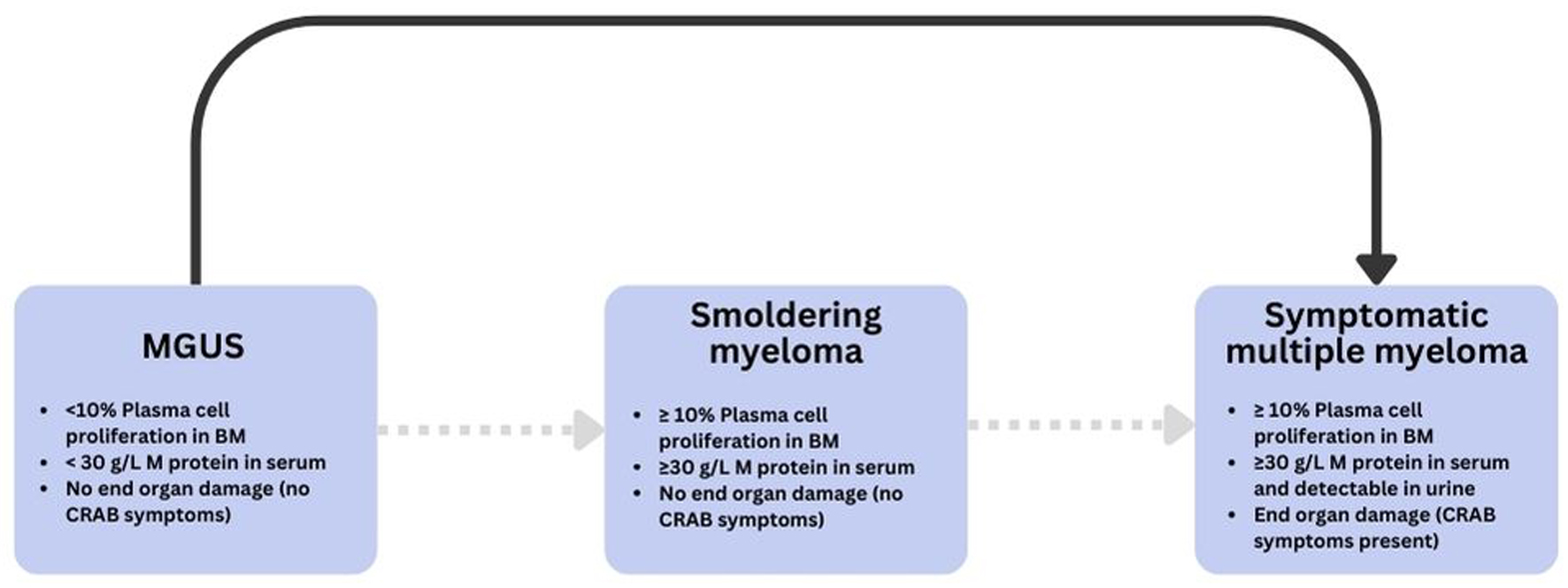 Click for large image | Figure 1. Progression and diagnostic criteria across the monoclonal gammopathy spectrum. Schematic overview of MGUS, smoldering myeloma, and symptomatic multiple myeloma with key thresholds (clonal plasma cells in bone marrow, serum M-protein) and CRAB/end-organ damage criteria [9]. BM: bone marrow; CRAB: hypercalcemia, renal insufficiency, anemia, bone lesions; MGUS: monoclonal gammopathy of uncertain significance. |
The underlying pathogenesis of MM is the result of the unregulated proliferation of antibody producing plasma cells, leading to the accumulation of high molecular weight proteins in the bone marrow and blood. Patients can present with the characteristic hypercalcemia, renal failure, anemia, bone lesions (CRAB) symptoms (Table 1) [9], in addition to symptoms of hyperviscosity syndrome such as bleeding, visual disturbances, and neurological deficits due to the accumulation of the heavy IgM proteins. Patients can also present with signs and symptoms that indicate bone marrow infiltration such as cytopenias and bone lesions [10]. The type of antibody secreted can result in a complex and heterogenous group of plasma cell disorders that include non-secretory MM, immunoglobulin G (IgG) MM, immunoglobulin A (IgA) MM, and the exceedingly rare, immunoglobulin M (IgM) MM [11]. IgM MM, also known as IgM myeloma, is a subtype of MM that constitutes 0.5-1% of myeloma cases [12]. The diagnosis of IgM myeloma is defined by IMWG and is based on the presence of IgM monoclonal protein in the serum and/or urine, along with evidence of bone marrow involvement by malignant plasma cells (Table 1) [10].
 Click to view | Table 1. Revised IMWG Diagnostic Criteria for MM (Adapted From IMWG Guidelines [9]) |
IgM MM has significant clinical challenges, with most data derived from small series and case reports. Additionally, distinguishing IgM myeloma from Waldenstrom macroglobulinemia (WM) remains a recurrent difficulty [12]. This review aims to provide a comprehensive synthesis of the current literature on its pathogenesis, clinical presentation, diagnostic criteria, and therapeutic strategies. We also highlight key distinctions between IgM myeloma and other IgM gammopathies, particularly WM, to aid in accurate diagnosis. Our objective is to consolidate available data for clinicians managing this rare subtype of MM.
| Methodology | ▴Top |
We conducted a comprehensive literature review using PubMed and Web of Science databases supplemented by referencing management software for article screening and citation management. Keywords including “IgM Multiple Myeloma”, “IgM Myeloma”, “Multiple myeloma”, “gammopathies”, and “monoclonal gammopathy” were employed resulting in 1,842 studies. Filters were applied to include only articles published in English, observational studies, systematic reviews, case reports, and case series within the past 10 years yielding 103 studies out of the originally obtained 1,842. Two independent reviewers (FE and RF) screened the 103 remaining articles based on screening of titles, abstracts, and full texts. Discrepancies between reviewers were resolved through discussion, and if consensus was not reached, a third reviewer (HE) served as adjudicator. A total of 49 studies were finally included (Table 2, Fig. 2).
Click to view | Table 2. Summary of Literature Review Process |
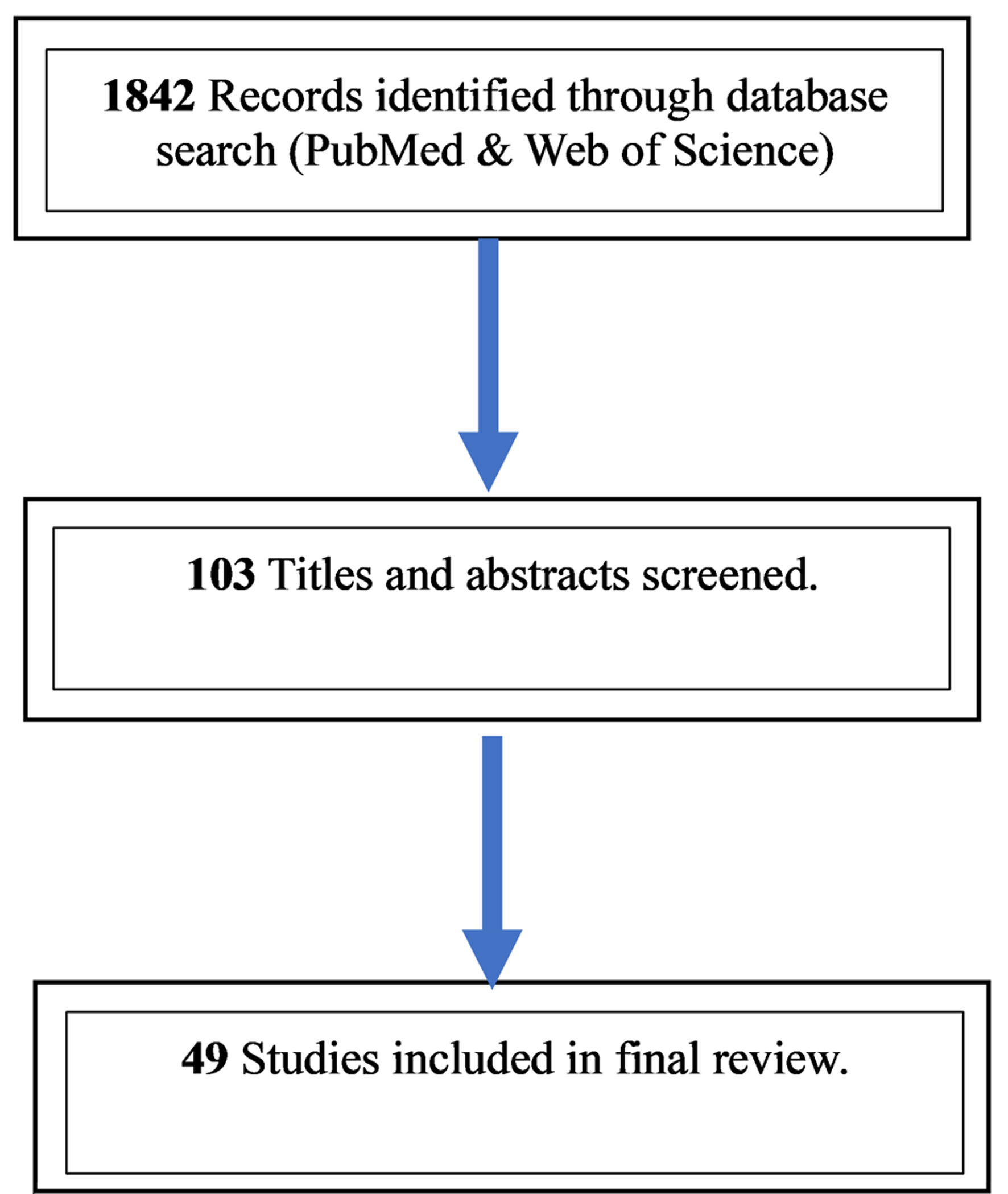 Click for large image | Figure 2. PRISMA-style study selection for the IgM myeloma review. Database searches of PubMed and Web of Science (English, human studies, last 10 years) identified 1,842 records; 103 full texts were assessed after limits/de-duplication; 49 studies were included in the qualitative synthesis. |
| Pathophysiology | ▴Top |
The most frequent karyotypic alteration in IgM myeloma involves translocations at the immunoglobulin heavy chain (IgH) locus, located at 14q32. This locus is highly transcriptionally active in B and plasma cells, so transferring an oncogene to 14q32 will cause dysregulation. Errors during class-switch recombination or somatic hypermutation can generate these IgH translocations [13].
In IgM myeloma, IgH translocations mirror those seen across MM, most commonly t(11;14)(q13;q32) with CCND1 activation, and less frequently t(4;14)(p16;q32) (involving FGFR3/NSD2) and t(14;16)(q32;q23) (MAF); rarer t(6;14) events may involve IRF4/MUM1 or CCND3. Across published cohorts, the prevalence of t(11;14) in IgM myeloma is about 40% in multicenter series and 60-80% in small, focused cohorts, reflecting methodological and population differences [13-19]. Immunophenotypically, IgM-myeloma plasma cells typically lose CD19, CD27, and CD45, with frequent aberrant expression of CD56, CD20, CD117, and cyclin D1, the latter two being enriched in t(11;14) disease. In the same series [18], involving 134 patients, CD20 expression was positive in 58% of the cases evaluated. Cyclin D1 expression was observed in approximately two-thirds of the cases. These features overlap with other MM isotypes; therefore, immunophenotyping should be interpreted alongside cytogenetics and clinical context (Fig. 3) [20].
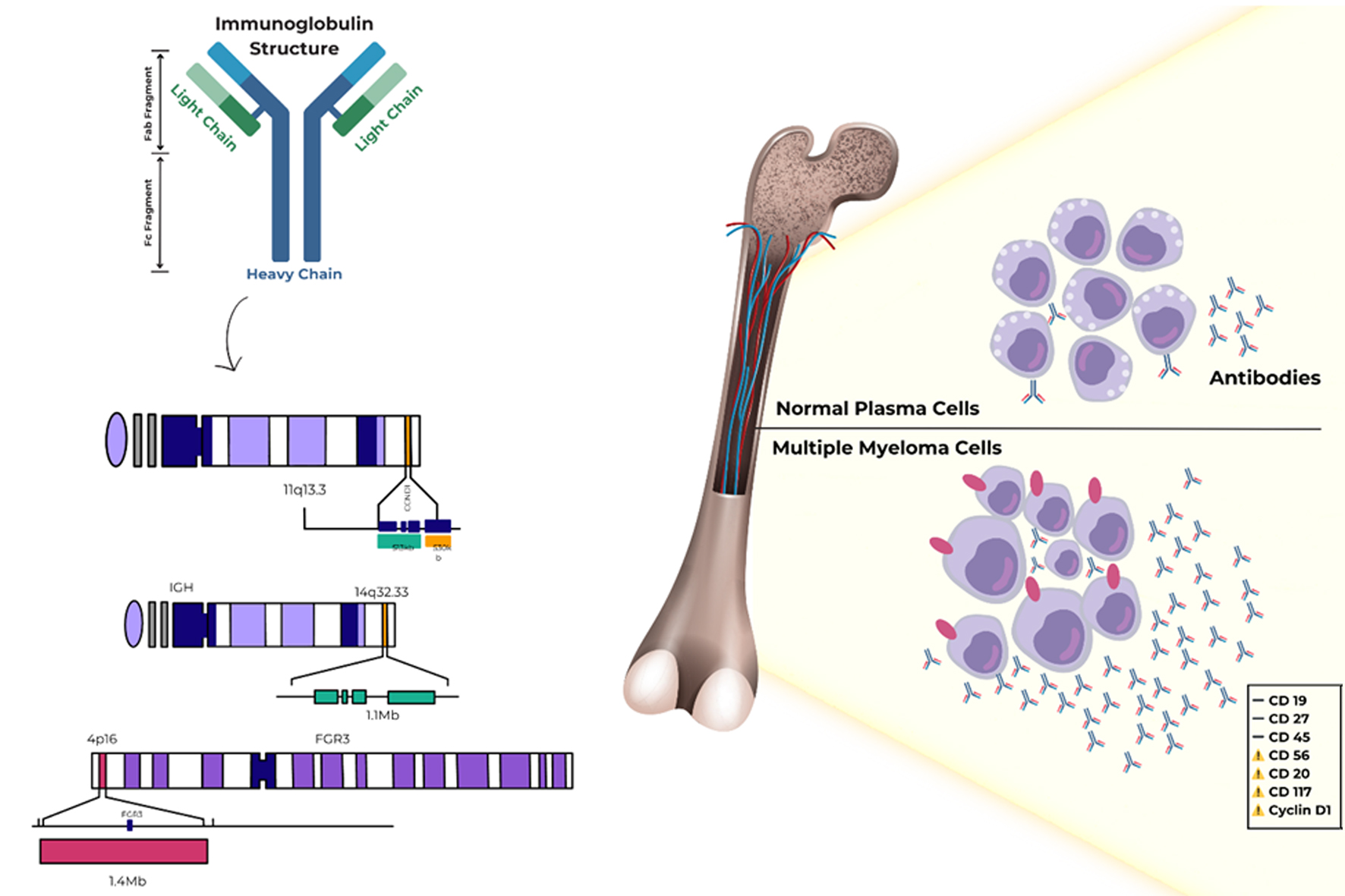 Click for large image | Figure 3. Immunophenotype and IGH translocations in IgM myeloma. Common IGH rearrangements at 14q32 (e.g., t(11;14) → CCND1, t(4;14) → FGFR3/NSD2, t(14;16) → MAF; rarer t(6;14) → IRF4/MUM1/CCND3) and representative immunophenotype of IgM myeloma plasma cells: loss of CD19/CD27/CD45 with aberrant CD56/CD20/CD117/cyclin D1. IGH: immunoglobulin heavy chain; WM: Waldenstrom macroglobulinemia. |
In contrast, WM shows a lymphoplasmacytic phenotype with retention of B-cell antigens (e.g., CD19, CD20) and a distinct genetic profile; critically, IgM myeloma is typically MYD88 L265P-negative, which supports its separation from WM (see section “Diagnostics”) [21, 22].
| Clinical Features | ▴Top |
The median age of presentation and diagnosis of IgM myeloma is reported to be 65 years, with a male-to-female ratio of 2:1. The typical myeloma symptoms such as hypercalcemia, renal failure, and anemia are equally common in IgM myeloma as it is in other types of myelomas [18, 23, 24]. Lytic bone findings are also seen with IgM myeloma and are part of the diagnostic criteria [25]. This was demonstrated in a retrospective study done on 134 patients, where the median age at diagnosis was 65.5 years. Males had a higher predominance than females (68%) and the typical myeloma features such as anemia, elevated serum calcium levels, renal dysfunction, and skeletal lytic lesions were found in 37%, 43%, 19%, and 70% of patients, respectively [18].
In the same study [18], hyperviscosity has been more commonly reported in IgM myeloma in comparison to the other myeloma types, which is likely due to the unique biophysical properties of IgM antibodies. IgM is a pentameric immunoglobulin with a molecular weight of about 970 kDa, significantly larger than IgG or IgA. This structural characteristic increases serum viscosity even at relatively modest monoclonal protein levels. In contrast to IgG MM, where viscosity usually rises only when paraprotein levels exceed 4 - 5 g/dL, patients with IgM paraprotein may develop symptomatic hyperviscosity (e.g., visual disturbances, mucosal bleeding, neurologic changes) at lower concentrations. This phenomenon is also prominent in WM but is mechanistically relevant to IgM MM as well, due to the same immunoglobulin class [26].
Additionally, acquired von Willebrand disease (aVWD) has been documented in IgM myeloma, likely caused by the adsorption and clearance of von Willebrand factor by the monoclonal IgM paraprotein, contributing to bleeding diatheses [27].
Organomegaly is a feature more commonly reported in other types of gammopathies, specifically WM. However, some IgM myeloma patients can also present with organomegaly as reported in Avet-Loiseau et al, where a prevalence of organomegaly in 25% (two out of eight) was reported [21]. Lymphadenopathy and hepatosplenomegaly may be present in some patients, as reported by De Gramont et al and Zarrabi et al, although these symptoms are typically more common in lymphoma than in myeloma [28, 29]. Histopathologically, IgM myeloma is characterized by a pure plasmocytic morphology, which differentiates it from WM, which has a lymphoplasmacytic morphology [17, 30].
| Diagnostics | ▴Top |
It is essential to distinguish IgM myeloma from other monoclonal gammopathies, as the management approach differs significantly from other types (Table 3) [18, 19, 30, 31].
Click to view | Table 3. Differential Diagnoses of IgM Myeloma |
The distinction between IgM myeloma and other IgM-related gammopathies, particularly WM is essential due to their divergent biology, clinical behavior, and treatment. IgM myeloma typically shows a pure plasmacytic morphology, lytic bone lesions, hypercalcemia, renal dysfunction, and the absence of organomegaly, features that are rare in WM. Immunophenotypically, IgM myeloma frequently lacks normal B-cell markers such as CD19 and CD20 and instead exhibits aberrant expression of CD56 and cyclin D1, especially in the presence of the t(11;14) translocation. In contrast, WM characteristically retains B-cell markers and has a lymphoplasmacytic morphology. A pivotal molecular distinction is the absence of the MYD88 L265P mutation in IgM myeloma, which is present in over 90% of WM cases. This mutation serves as a critical biomarker that helps differentiate between the two entities in diagnostically challenging cases. Additionally, WM often presents with organomegaly and hyperviscosity, whereas IgM myeloma is more commonly associated with skeletal involvement and CRAB symptoms [19, 31].
In addition to plasma-cell IgM myeloma and WM, IgM gammopathies include IgM MGUS and IgM MGCS. IgM MGUS is defined by an asymptomatic circulating IgM M-protein < 30 g/L with < 10% lymphoplasmacytic marrow infiltration. Its natural history is indolent, with a progression risk of about 1.1 events per 100 person-years (most commonly to lymphoma/chronic lymphocytic leukemia (CLL)/AL amyloidosis); notably, in the largest series (n = 210), no patients progressed to plasma-cell IgM myeloma. IgM MGCS denotes IgM-mediated organ damage (e.g., neuropathy, cryoglobulinemia, cold agglutinin disease (CAD)) without meeting criteria for WM or myeloma; management targets the IgM-driven pathology [32].
To establish a definitive diagnosis, bone marrow morphology and immunophenotypic analysis are essential to establish a definitive diagnosis. The diagnostic workup typically begins by taking a focused history and physical examination, looking for possible features of organomegaly, a history of bone pain, and features of amyloidosis such as heart failure, chronic diarrhea, and orthostatic hypotension. Consequently, specific tests are conducted to help in ruling out differential diagnoses for IgM myeloma such as WM, IgM MGUS, IgM amyloidosis, and IgM-related disorders such as IgM neuropathies, cryoglobulinemia (type I and type II), CAD, polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes (POEMS) syndrome, Schnitzler syndrome, pyoderma gangrenous, scleromyxedema, and monoclonal gammopathy of renal significance [19, 30, 33-37].
Electrophoresis is used as an initial screening test in patients with suspected myelomas. After establishing a possible diagnosis of a monoclonal gammopathy, unilateral bone marrow aspirate and biopsy, including immunohistochemistry and/or flow cytometry, and cytogenetics are done next. Fluorescence in situ hybridization (FISH) is often utilized to reveal chromosomal abnormalities such as the translocations described before. Immunohistochemistry is also used to detect myeloma surface cell markers, which helps in differentiating IgM myeloma from other gammopathies [38]. When evaluating patients with suspected IgM-related disorders, special attention must be paid to cryoglobulin detection, which requires samples to be taken in pre-warmed tubes and maintained at 37 °C until serum separation to prevent precipitation and false-negative results. Even minimal amounts of measurable cryoglobulin may result in symptoms, and repeat testing is indicated if clinical suspicion remains high [32].
Since most MM patients have bone involvement, imaging techniques such as skeletal bone survey are considered to be the standard diagnostic approach in diagnosing lytic bone disease in MM [39]. However, skeletal bone surveys have their own limitations; in order for it to detect osteolytic lesions, 30% of the bone cortex should have been destroyed by the time of diagnosis [40].18F-FDG-PET/CT, multidetector computed tomography (MDCT), and low-dose computed tomography (LDCT) have shown to be superior in diagnosing MM bone disease than skeletal bone surveys [41-43].
The diagnosis of IgM myeloma currently requires the detection of IgM monoclonal gammopathy, at least 10% plasma cells in a bone marrow biopsy, and the presence of lytic bone lesions and/or the t(11;14) translocation identified through FISH [18, 39]. Additionally, the expression of cyclin D1, the presence of the t(11;14) translocation, and the absence of the MYD88 L265P gene mutation can aid in distinguishing IgM myeloma from WM [18].
| Prognosis | ▴Top |
The International Staging System (ISS) is a staging system established by the IMWG in 2005 to classify MM based on several prognostic factors (Table 4) [39, 44].
Click to view | Table 4. ISS for Multiple Myeloma and Estimated Median Survival |
The ISS is based on two variables, serum albumin and β2-microglobulin, which have proved to be reliable predictive factors for survival. This system was developed in North America, Europe, and Asia, in patients younger and older than age 65 years, and with standard therapy or auto transplant where it continued to display effectiveness [44]. This system was compared to the Durie-Salmon Staging System, which was developed in 1975, and it was found to remain effective [45]. A revised version of the ISS was then introduced in 2015, and it incorporated additional risk stratifiers such as cytogenetic abnormalities and lactate dehydrogenase (LDH) [44]. The prognosis of IgM myeloma specifically depends on the ISS stage and the clinical condition of the patient [46]. Castillo et al reported a more ominous prognosis in IgM myeloma compared to other types of myelomas. The same study suggested that factors like advanced age, female sex, and a high ISS may be associated with worse outcomes, even though there are limited data on whether IgM myeloma has disease-specific prognostic factors compared to other monoclonal gammopathies. Notably, patients classified as ISS stage III had a median survival of approximately 30 months, whereas those in stages I and II often lived more than 5 years [18].
| Therapeutic Management | ▴Top |
Due to the rarity of IgM myeloma, most therapeutic recommendations are extrapolated from studies of broader MM populations. Specific clinical trials exclusively evaluating IgM MM are lacking; thus, treatment strategies are adapted from the standard-of-care approaches used in other MM subtypes.
Front-line induction
Treatment strategies for IgM myeloma follow standard MM protocols with induction therapy consisting of triple or quadruple therapy (steroids, proteasome inhibitors, immunomodulators, monoclonal antibodies), followed by hematopoietic stem cell transplant depending on cytogenetic findings and transplant eligibility, then maintenance therapy [18, 22]. This management differs from WM management, which is based on the symptoms and depends on emergent plasmapheresis for hyperviscosity when IgM levels exceed 4,000 mg/dL, along with rituximab-based therapies or Bruton’s tyrosine kinase (BTK) inhibitors as first-line treatment [22].
Immunomodulatory drugs (IMiDs)
IMiDs, like thalidomide (thal) and lenalidomide (len), have demonstrated efficacy in treating IgM myeloma. Both thalidomide and its structural derivative, lenalidomide, are used to treat various illnesses. They exert their antimyeloma activity through multiple, now partially elucidated mechanisms. A key pathway involves binding to the cereblon (CRBN) E3 ubiquitin ligase complex, leading to targeted degradation of transcription factors Ikaros (IKZF1) and Aiolos (IKZF3), which are critical for myeloma cell survival. Additionally, IMiDs enhance T-cell and natural killer (NK) cell activation, inhibit regulatory T cells, suppress pro-inflammatory cytokine production, and exert anti-angiogenic effects within the tumor microenvironment [47, 48].
Lenalidomide plus dexamethasone has shown deep and durable responses in non-IgM MM, achieving ≥ 50% serum M-protein reduction in approximately 90% of patients in early and larger trials [49, 50]. Although direct data in IgM myeloma are limited, retrospective studies suggest similar benefit in this subgroup [18].
Moreover, lenalidomide and thalidomide’s ability to boost interleukin (IL)-2 production and suppress IgM secretion supports their rationale for use in IgM myeloma. The trend is moving towards substituting thalidomide/dexamethasone with lenalidomide/dexamethasone, with some guidelines even proposing lenalidomide monotherapy [51, 52].
Anti-CD38 monoclonal antibodies
Daratumumab, a monoclonal antibody targeting CD38, has become a significant treatment option for resistant or relapsed MM. Recent data from large-scale phase III trials have reshaped the first-line therapy for transplant-eligible and ineligible MM patients. The PERSEUS trial demonstrated that adding subcutaneous daratumumab to VRd (D-VRd) significantly improved progression-free survival (PFS), depth of response (complete response or better), and minimal residual disease (MRD) negativity rates compared to VRd alone. The estimated 48-month PFS rates were 84.3% for D-VRd versus 67.7% for VRd [53]. Additionally, the CEPHEUS trial demonstrated that the D-VRd regimen also led to a significantly deeper and more durable increase in MRD negativity, higher rates of complete response or better, and a 43% lower risk of progression or death compared to VRd alone [54]. Consequently, quadruplet therapy is now increasingly adopted as the global standard of care for newly diagnosed MM. While these data are not specific to IgM myeloma, the principles are likely applicable and support the inclusion of CD38-targeted therapies in front-line regimens when available.
Relapsed/refractory disease and targeted therapy (t(11;14))
In the context of innovative treatments, venetoclax has become the first approved selective B-cell lymphoma 2 (Bcl-2) inhibitor in its class, particularly beneficial for MM patients carrying the t(11;14) (q13; q32) translocation. Venetoclax monotherapy has shown effectiveness in treating relapsed/refractory multiple myeloma (RRMM). However, combining venetoclax with dexamethasone, in the presence or absence of bortezomib, has demonstrated even greater efficacy [55, 56]. The BELLINI trial, a phase III study assessing venetoclax in combination with bortezomib and dexamethasone, demonstrated improved PFS, particularly in patients harboring the t(11;14) translocation. However, an unexpected increase in treatment-related mortality, primarily due to infections in the overall study population, prompted the FDA to place a partial clinical hold on venetoclax trials in MM. Subsequent analyses clarified that this increased mortality was not observed in the t(11;14) subgroup, who derived the most benefit and had acceptable safety outcomes. Accordingly, venetoclax use in MM, including IgM MM, should be restricted to carefully selected t(11;14) patients, with attention to infection risk [56, 57]. Case experiences with venetoclax/carfilzomib/dexamethasone (VenKD) show rapid responses but relapses in some individuals within months [58].
T-cell-redirecting therapies
Historically labeled as “novel agents,” bortezomib, lenalidomide, and thalidomide revolutionized multiple myeloma therapy in the early 2000s. However, this terminology has become outdated given the emergence of more recent immunotherapies that have substantially reshaped the treatment landscape. These include bispecific T-cell engagers such as teclistamab, talquetamab, and elranatamab, as well as chimeric antigen receptor (CAR) T-cell therapies like idecabtagene vicleucel and ciltacabtagene autoleucel, which are now incorporated into international treatment guidelines for RRMM. These agents have demonstrated high response rates in heavily pretreated patients and are increasingly considered earlier in the treatment algorithm [59-61]. Bispecific antibodies such as teclistamab (targeting BCMA and CD3) have shown response rates of about 63% in patients with RRMM [59], while CAR T-cell therapies like idecabtagene vicleucel and ciltacabtagene autoleucel have demonstrated overall response rates of 70-97%, including durable remissions in some cases [62, 63]. However, their application to rare subtypes like IgM myeloma remains largely theoretical due to a lack of subtype-specific data.
Autologous stem cell transplantation (ASCT)
In transplant-eligible patients, the contemporary standard mirrors other MM isotypes: induction therapy followed by ASCT and post-transplant maintenance. Induction typically comprises triplet or quadruplet regimens such as bortezomib, lenalidomide, and dexamethasone (VRd), or combinations incorporating monoclonal antibodies like daratumumab. Indeed, daratumumab-based quadruplets have become front-line standard-of-care regimens in many parts of the world, including both standard- and high-risk patients, as supported by the CASSIOPEIA (D-VTd), GRIFFIN (D-VRd), and MAIA (D-Rd in transplant-ineligible) trials [64-66]. In our setting, the availability and reimbursement of daratumumab may currently limit its use primarily to high-risk patients or those with relapsed/refractory disease. Nonetheless, we acknowledge its global role in first-line treatment and encourage its integration as access improves.
Regarding ASCT, it remains the cornerstone of therapy for eligible patients and is associated with improved PFS. While transplant-related risks exist, they are generally low in contemporary practice (< 1-2%) and outweighed by the survival benefits conferred in properly selected patients. Long-term outcomes are optimized when patients achieve deep responses prior to transplant, and post-ASCT maintenance further enhances durability [67]. Factors influencing ASCT success include patient-specific and biological variables, such as baseline albumin, β2-microglobulin levels, and chemotherapy sensitivity [68, 69]. The type of M-protein present at transplantation is a reliable indicator of the effectiveness of initial chemotherapy, impacting the likelihood of achieving a complete remission after ASCT [70]. Some studies propose that double transplantation may be more beneficial in treating patients with MM or preventing relapses compared to single transplantation [71]. Following ASCT, patients experience a good quality of life, with some requiring only maintenance therapy rather than sequential chemotherapy [72]. The DETERMINATION trial, a randomized trial, focuses on the importance of individualizing the management strategies for MM patients. It shows that VRd therapy followed by early ASCT provides a PFS advantage; however, overall survival rates do not significantly differ whether ASCT is performed early or delayed. This concludes that the management should be tailored according to several factors including patient preferences and disease characteristics [73]. Notably, the last 15 to 20 years have seen significant advancements in treating IgM myeloma. The current protocol for transplant-eligible patients includes induction, stem cell mobilization, and ASCT, followed by maintenance or consolidation [72].
Historical outcomes context
Earlier series reported limited complete responses with conventional chemotherapy and a median survival of about 3 years in IgM MM [68]. In a cohort transplanted between 1997 and 2006 following ≥ 2 chemotherapy cycles (n = 122; including one IgM case among predominantly IgG/IgA subtypes), resistance to older regimens supported high-dose therapy with ASCT as the standard for fit patients < 65 years with adequate organ function [68, 74].
| Supportive Management | ▴Top |
Supportive strategies for IgM myeloma mirror standard MM practice; major guidelines do not distinguish by heavy-chain isotype. Care should include risk-adapted infection prophylaxis, structured skeletal management, evidence-based analgesia, and targeted treatment of anemia [75].
Infections remain a major driver of early morbidity and mortality in MM, with the highest risk in the first months after starting therapy. However, there are no IgM-specific data, so recommendations are extrapolated from general MM guidance [75]. For newly diagnosed patients beginning induction, a time-limited 12-week course of oral levofloxacin can be considered to reduce febrile episodes and early deaths, with no routine extension beyond 12 weeks (TEAMM trial) [76]. Antiviral prophylaxis (acyclovir/valacyclovir) is recommended for patients receiving proteasome inhibitors or anti-CD38 antibodies and routinely around ASCT [77, 78]. Pneumocystis prophylaxis (e.g., trimethoprim-sulfamethoxazole (TMP-SMX)) is reserved for higher-risk settings such as prolonged high-dose corticosteroids, intensive therapy, or the early post-ASCT period [79]. Seasonal influenza and pneumococcal vaccinations should be administered per schedules. IVIG is not routine but may be used in selected patients with recurrent/severe infections and marked hypogammaglobulinemia [75]. Collectively, these measures should be applied to IgM myeloma with the same risk-adapted approach used in other MM isotypes.
In MM, the unregulated growth of malignant plasma cells in the bone marrow leads to osteolysis and bone loss, which significantly increases morbidity. Several studies have shown the importance of bisphosphonates for palliative care due to their effectiveness in lowering the risk of hypercalcemia and skeletal complications associated with myeloma. The IMWG Bone Working Group recommends zoledronic acid (or pamidronate) for all patients with active MM, regardless of baseline lytic lesions, with renal-adjusted dosing and mandatory dental evaluation and calcium/vitamin D supplementation. After about 12 months, dosing can be de-escalated (e.g., every 3 months) in deep responders, with continuation or re-initiation at biochemical/clinical relapse [80, 81].
Analgesia should follow cancer-pain guidelines: use regular non-opioids and strong opioids for moderate-severe nociceptive pain, titrated with breakthrough dosing and functional reassessment. For neuropathic components, adjuvant agents such as gabapentinoids or duloxetine can be added; randomized data support duloxetine for chemotherapy-induced painful neuropathy. Non-pharmacologic and interventional measures (e.g., radiotherapy, vertebral augmentation) can be used when indicated [82-84].
Anemia is a common complication in MM, including IgM MM, resulting from marrow infiltration, inflammation-driven hepcidin-mediated iron restriction, relative erythropoietin deficiency from renal dysfunction, and treatment-related myelosuppression [85]. Erythropoiesis-stimulating agents (ESAs, epoetin alfa or darbepoetin alfa) may be considered for chemotherapy-associated, symptomatic anemia in non-curative settings to raise hemoglobin (Hb) and reduce transfusions; however, they require risk-benefit discussion given increased venous thromboembolism risk and signals of higher mortality in meta-analyses [86, 87]. Current guidelines recommend ESAs selectively (typically when Hb < 10 g/dL), aiming for the lowest Hb sufficient to avoid transfusion, and after correcting iron deficiency [86]. Adding intravenous iron improves ESA response and further lowers transfusion needs [88]. For hospitalized, hemodynamically stable hematology-oncology patients, a restrictive red blood cell (RBC) transfusion threshold around 7 g/dL is recommended, individualized to symptoms/comorbidity [89]. There are no IgM-specific trials; practice is extrapolated from general MM/oncology data [90, 91].
Given the limited body of literature discussing IgM myeloma, most of the management guidance in this review is extrapolated by data from broader MM populations. Wherever possible, we have cited IgM-specific findings, but for certain therapeutic strategies and prognostic frameworks, extrapolation from general MM studies remains necessary. We highlight the urgent need for dedicated prospective studies focused on this rare subtype.
| Conclusion | ▴Top |
IgM myeloma is reported to have a more ominous prognosis compared to the remaining MM subtypes in some cases. This places heavy emphasis on timely diagnosis and effective management. Current diagnostics rely on a combination of clinical assessment, laboratory investigations, bone marrow biopsies, cytogenetics, and immunohistochemistry studies. Following diagnosis, appropriate management is imperative to prevent the progression of the disorder. The current treatment options for IgM myeloma are primarily adapted from those used for other types of MM, but there is a need for more specific and tailored therapies. We invite future studies that investigate the efficacy of the current treatments of IgM myeloma and the development of standardized guidelines for the management of this rare subtype of MM.
Acknowledgments
None to declare.
Financial Disclosure
No funding was received.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
Hadeel Elwaheidi: conceptualization, literature search, data curation, manuscript drafting, and critical revisions. Alaa Hamad: literature search, writing - original draft, figure preparation, and reference management. Farah Salameh: data extraction, writing - review and editing, and formatting. Fadwa Elkordy: draft refinement, table construction, and proofreading. Rojina FathAlrahman: writing - background and clinical features sections, and figure design. Amr Hanbali: supervision, expert review of hematologic content, and final approval of the manuscript. All authors have read and approved the final version of the manuscript.
Data Availability
The data supporting the conclusions of this review article are available from the cited references.
Abbreviations
ASCT: autologous stem cell transplantation; Bcl-2: B-cell lymphoma 2; CD: cluster of differentiation; CRAB: hypercalcemia, renal failure, anemia, bone lesions; D-VRd: daratumumab, bortezomib, lenalidomide, and dexamethasone regimen; FISH: fluorescence in situ hybridization; IgM: immunoglobulin M; ISS: International Staging System; LDCT: low-dose computed tomography; MGUS: monoclonal gammopathy of undetermined significance; MM: multiple myeloma; MRI: magnetic resonance imaging; PET-CT: positron emission tomography-computed tomography; Sβ2M: serum beta-2 microglobulin; t(11;14): translocation between chromosomes 11 and 14; VRd: bortezomib, lenalidomide, and dexamethasone regimen; WM: Waldenstrom’s macroglobulinemia
| References | ▴Top |
- Assessment UENC for E. WHO classification of tumours of haematopoietic and lymphoid tissues. 2009.
- Lu H, Durkin L, Zhao X, Nakashima MO. IgM plasma cell myeloma: clinicopathologic features including evaluation for MYD88 and CXCR4 mutations. Am J Clin Pathol. 2022;157(1):47-53.
doi pubmed - Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004;351(18):1860-1873.
doi pubmed - Ludwig H, Novis Durie S, Meckl A, Hinke A, Durie B. Multiple myeloma incidence and mortality around the globe; Interrelations Between Health Access and Quality, Economic Resources, and Patient Empowerment. Oncologist. 2020;25(9):e1406-e1413.
doi pubmed - Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, Hayes RB, Dispenzieri A, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113(22):5412-5417.
doi pubmed - Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Offord JR, Dispenzieri A, et al. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med. 2006;354(13):1362-1369.
doi pubmed - Fairfield H, Falank C, Avery L, Reagan MR. Multiple myeloma in the marrow: pathogenesis and treatments. Ann N Y Acad Sci. 2016;1364(1):32-51.
doi pubmed - Rajkumar SV, Landgren O, Mateos MV. Smoldering multiple myeloma. Blood. 2015;125(20):3069-3075.
doi pubmed - International Myeloma Working G. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121(5):749-757.
pubmed - Girard LP, Soekojo CY, Ooi M, Poon LM, Chng WJ, de Mel S. Immunoglobulin M paraproteinaemias. Cancers (Basel). 2020;12(6):1-21.
doi pubmed - Greuter T, Browne M, Dommann-Scherrer C, Binder D, Renner C, Kapp U. IgM multiple myeloma with an extremely rare non-aggressive presentation: A case report. Oncol Lett. 2016;12(4):2801-2803.
doi pubmed - Hanbali A, Alamer A, Alhayli S. Uncommon entities, uncommon challenges: a review of rare plasma cell disorders. Hematol Rep. 2025;17(4):31.
doi pubmed - Fenton JA, Pratt G, Rawstron AC, Morgan GJ. Isotype class switching and the pathogenesis of multiple myeloma. Hematol Oncol. 2002;20(2):75-85.
doi pubmed - Feyler S, O'Connor SJ, Rawstron AC, Subash C, Ross FM, Pratt G, Drayson MT, et al. IgM myeloma: a rare entity characterized by a CD20-CD56-CD117- immunophenotype and the t(11;14). Br J Haematol. 2008;140(5):547-551.
doi pubmed - Avet-Loiseau H, Garand R, Lode L, Harousseau JL, Bataille R, Intergroupe Francophone du M. Translocation t(11;14)(q13;q32) is the hallmark of IgM, IgE, and nonsecretory multiple myeloma variants. Blood. 2003;101(4):1570-1571.
doi pubmed - Ackroyd S, O'Connor SJ, Rawstron AC, Owen RG. IgM myeloma with t(4;14)(p16;q32). Cancer Genet Cytogenet. 2005;162(2):183-184.
doi pubmed - King RL, Howard MT, Hodnefield JM, Morice WG. IgM multiple myeloma: pathologic evaluation of a rare entity. Am J Clin Pathol. 2013;140(4):519-524.
doi pubmed - Castillo JJ, Jurczyszyn A, Brozova L, Crusoe E, Czepiel J, Davila J, Dispenzieri A, et al. IgM myeloma: a multicenter retrospective study of 134 patients. Am J Hematol. 2017;92(8):746-751.
doi pubmed - Schuster SR, Rajkumar SV, Dispenzieri A, Morice W, Aspitia AM, Ansell S, Kyle R, et al. IgM multiple myeloma: disease definition, prognosis, and differentiation from Waldenstrom's macroglobulinemia. Am J Hematol. 2010;85(11):853-855.
doi pubmed - Burns GF, Cawley JC, Barker CR, Worman CP, Raper CG, Hayhoe FG. Differing surface marker characteristics in plasma cell dyscrasias with particular reference to IgM myeloma. Clin Exp Immunol. 1978;31(3):414-418.
pubmed - Avet-Loiseau H, Garand R, Lode L, Robillard N, Bataille R. 14q32 Translocations discriminate IgM multiple myeloma from Waldenstrom's macroglobulinemia. Semin Oncol. 2003;30(2):153-155.
doi pubmed - Joseph K, Greidinger A, Koch M. IgM Multiple myeloma: a rare clinical entity and diagnostic dilemma. Cooper Rowan Med J [Internet]. 2021;3(1):2578-3343. Available from: https://rdw.rowan.edu/crjcsm/vol3/iss1/6CooperRowanMedicalJournal:https://rdw.rowan.edu/crjcsm.
- Tathineni P, Cancarevic I, Malik BH. Uncommon presentations of multiple myeloma. Cureus. 2020;12(6):e8400.
doi pubmed - Zetter DR, Kabir T, Reynolds SB. The clinical characteristics of multiple myeloma in the acute care setting: case presentation and clinical recommendations. Cureus. 2020;12(9):e10463.
doi pubmed - Donovan KA, Lacy MQ, Gertz MA, Lust JA. IL-1beta expression in IgM monoclonal gammopathy and its relationship to multiple myeloma. Leukemia. 2002;16(3):382-385.
doi pubmed - Mehta J, Singhal S. Hyperviscosity syndrome in plasma cell dyscrasias. Semin Thromb Hemost. 2003;29(5):467-471.
doi pubmed - Hopting M, Budde U, Tiede A, Grube M, Hahn J, Herr W, Heimerl S, et al. Distinct mechanisms of IgM antibody-mediated acquired von willebrand syndrome and successful treatment with recombinant von Willebrand factor in one patient. Acta Haematol. 2022;145(4):454-457.
doi pubmed - Zarrabi MH, Stark RS, Kane P, Dannaher CL, Chandor S. IgM myeloma, a distinct entity in the spectrum of B-cell neoplasia. Am J Clin Pathol. 1981;75(1):1-10.
doi pubmed - De Gramont A, Grosbois B, Michaux JL, Peny AM, Pollet JP, Smadja N, Krulik M, et al. [IgM myeloma: 6 cases and a review of the literature]. Rev Med Interne. 1990;11(1):13-18.
doi pubmed - Owen RG, Treon SP, Al-Katib A, Fonseca R, Greipp PR, McMaster ML, Morra E, et al. Clinicopathological definition of Waldenstrom's macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom's Macroglobulinemia. Semin Oncol. 2003;30(2):110-115.
doi pubmed - Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, Sheehy P, et al. MYD88 L265P somatic mutation in Waldenstrom's macroglobulinemia. N Engl J Med. 2012;367(9):826-833.
doi pubmed - Khwaja J, Salter SJ, D’Sa S. IgM-associated cryoglobulinaemia. Hemato. 2023 Jul 21;4(3):240-249.
- Bhatt VR, Murukutla S, Naqi M, Pant S, Kedia S, Terjanian T. IgM myeloma or Waldenstrom's macroglobulinemia is the big question? Maedica (Bucur). 2014;9(1):72-75.
pubmed - Nordquist LT, Saba HI, Moscinski LC. IgM myeloma: an IgM gammopathy distinct from Waldenstrom's macroglobulinemia. Hematology. 2001;6(1):53-58.
doi pubmed - Chehal A, Taher A, Shamseddine A. IgM myeloma and Waldenstrom's macroglobulinemia: a distinct clinical feature, histology, immunophenotype, and chromosomal abnormality. Clin Lab Haematol. 2003;25(3):187-190.
doi pubmed - Takahashi K, Yamamura F, Motoyama H. IgM myeloma—its distinction from Waldenstrom's macroglobulinemia. Acta Pathol Jpn. 1986;36(10):1553-1563.
doi pubmed - Tedeschi A, Conticello C, Rizzi R, Benevolo G, Laurenti L, Petrucci MT, Zaja F, et al. Diagnostic framing of IgM monoclonal gammopathy: focus on Waldenstrom macroglobulinemia. Hematol Oncol. 2019;37(2):117-128.
doi pubmed - Grunenberg A, Buske C. Monoclonal IgM gammopathy and Waldenstrom's macroglobulinemia. Dtsch Arztebl Int. 2017;114(44):745-751.
doi pubmed - Dimopoulos M, Terpos E, Comenzo RL, Tosi P, Beksac M, Sezer O, Siegel D, et al. International myeloma working group consensus statement and guidelines regarding the current role of imaging techniques in the diagnosis and monitoring of multiple Myeloma. Leukemia. 2009;23(9):1545-1556.
doi pubmed - Edelstyn GA, Gillespie PJ, Grebbell FS. The radiological demonstration of osseous metastases. Experimental observations. Clin Radiol. 1967;18(2):158-162.
doi pubmed - Breyer RJ, 3rd, Mulligan ME, Smith SE, Line BR, Badros AZ. Comparison of imaging with FDG PET/CT with other imaging modalities in myeloma. Skeletal Radiol. 2006;35(9):632-640.
doi pubmed - Mahnken AH, Wildberger JE, Gehbauer G, Schmitz-Rode T, Blaum M, Fabry U, Gunther RW. Multidetector CT of the spine in multiple myeloma: comparison with MR imaging and radiography. AJR Am J Roentgenol. 2002;178(6):1429-1436.
doi pubmed - Gleeson TG, Moriarty J, Shortt CP, Gleeson JP, Fitzpatrick P, Byrne B, McHugh J, et al. Accuracy of whole-body low-dose multidetector CT (WBLDCT) versus skeletal survey in the detection of myelomatous lesions, and correlation of disease distribution with whole-body MRI (WBMRI). Skeletal Radiol. 2009;38(3):225-236.
doi pubmed - Palumbo A, Avet-Loiseau H, Oliva S, Lokhorst HM, Goldschmidt H, Rosinol L, Richardson P, et al. Revised international staging system for multiple myeloma: a report from international myeloma working group. J Clin Oncol. 2015;33(26):2863-2869.
doi pubmed - Durie-Salmon Staging System | Int’l Myeloma Foundation [Internet]. [cited Mar 25, 2024]. Available from: https://www.myeloma.org/durie-salmon-staging.
- Dierlamm T, Laack E, Dierlamm J, Fiedler W, Hossfeld DK. IgM myeloma: a report of four cases. Ann Hematol. 2002;81(3):136-139.
doi pubmed - Kronke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, Chamberlain PP, et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature. 2015;523(7559):183-188.
doi pubmed - Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J, Karasawa S, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26(11):2326-2335.
doi pubmed - Rajkumar SV, Hayman SR, Lacy MQ, Dispenzieri A, Geyer SM, Kabat B, Zeldenrust SR, et al. Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood. 2005;106(13):4050-4053.
doi pubmed - Weber DM, Chen C, Niesvizky R, Wang M, Belch A, Stadtmauer EA, Siegel D, et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med. 2007;357(21):2133-2142.
doi pubmed - Richardson P, Hideshima T, Anderson K. Thalidomide: emerging role in cancer medicine. Annu Rev Med. 2002;53:629-657.
doi pubmed - Shannon E, Sandoval F, Greig N, Stagg P. Lenalidomide alone or lenalidomide plus dexamethasone significantly inhibit IgG and IgM in vitro… A possible explanation for their mechanism of action in treating multiple myeloma. Int Immunopharmacol. 2012;12(2):441-446.
doi pubmed - Sonneveld P, Dimopoulos MA, Boccadoro M, Quach H, Ho PJ, Beksac M, et al. Phase 3 randomized study of daratumumab (DARA) + Bortezomib, Lenalidomide, and Dexamethasone (VRd) versus Vrd alone in patients (Pts) with newly diagnosed multiple myeloma (NDMM) who are eligible for autologous stem cell transplantation (ASCT): primary results of the perseus trial. Blood. 2023;142(Supplement 2):LBA-1-LBA-1.
- Usmani SZ, Facon T, Hungria V, Bahlis NJ, Venner CP, Braunstein M, Pour L, et al. Daratumumab plus bortezomib, lenalidomide and dexamethasone for transplant-ineligible or transplant-deferred newly diagnosed multiple myeloma: the randomized phase 3 CEPHEUS trial. Nat Med. 2025;31(4):1195-1202.
doi pubmed - Moreau P, Chanan-Khan A, Roberts AW, Agarwal AB, Facon T, Kumar S, Touzeau C, et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood. 2017;130(22):2392-2400.
doi pubmed - Kumar S, Kaufman JL, Gasparetto C, Mikhael J, Vij R, Pegourie B, Benboubker L, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood. 2017;130(22):2401-2409.
doi pubmed - Kumar SK, Harrison SJ, Cavo M, de la Rubia J, Popat R, Gasparetto C, Hungria V, et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2020;21(12):1630-1642.
doi pubmed - Abuelgasim KA, Alherz N, Alhejazi A, Damlaj M. Venetoclax in combination with carfilzomib and dexamethasone in relapsed/refractory multiple myeloma harboring t(11,14)(q13;q32): two case reports and a review of the literature. J Med Case Rep. 2020;14(1):54.
doi pubmed - Moreau P, Garfall AL, van de Donk N, Nahi H, San-Miguel JF, Oriol A, Nooka AK, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. 2022;387(6):495-505.
doi pubmed - Firestone R, Lesokhin AM, Usmani SZ. An embarrassment of riches: three FDA-approved bispecific antibodies for relapsed refractory multiple myeloma. Blood Cancer Discov. 2023;4(6):433-436.
doi pubmed - Chari A, Touzeau C, Schinke C, Minnema MC, Berdeja JG, Oriol A, van de Donk N, et al. Safety and activity of talquetamab in patients with relapsed or refractory multiple myeloma (MonumenTAL-1): a multicentre, open-label, phase 1-2 study. Lancet Haematol. 2025;12(4):e269-e281.
doi pubmed - Munshi NC, Anderson LD, Jr., Shah N, Madduri D, Berdeja J, Lonial S, Raje N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705-716.
doi pubmed - Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, Stewart AK, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. 2021;398(10297):314-324.
doi pubmed - Moreau P, Attal M, Hulin C, Arnulf B, Belhadj K, Benboubker L, Bene MC, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet. 2019;394(10192):29-38.
doi pubmed - Voorhees PM, Kaufman JL, Laubach J, Sborov DW, Reeves B, Rodriguez C, Chari A, et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: the GRIFFIN trial. Blood. 2020;136(8):936-945.
doi pubmed - Facon T, Kumar S, Plesner T, Orlowski RZ, Moreau P, Bahlis N, Basu S, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380(22):2104-2115.
doi pubmed - Attal M, Lauwers-Cances V, Marit G, Caillot D, Moreau P, Facon T, Stoppa AM, et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366(19):1782-1791.
doi pubmed - Wach M, Cioch M, Hus M, Jawniak D, Legiec W, Malek M, Manko J, et al. Treatment of multiple myeloma patients with autologous stem cell transplantation - a fresh analysis. Folia Histochem Cytobiol. 2011;49(2):248-254.
doi pubmed - Attal M, Moreau P, Avet-Loiseau H, Harousseau JL. Stem cell transplantation in multiple myeloma. Hematology Am Soc Hematol Educ Program. 2007;2007(1):311-316.
doi pubmed - Alexanian R, Weber D, Giralt S, Dimopoulos M, Delasalle K, Smith T, Champlin R. Impact of complete remission with intensive therapy in patients with responsive multiple myeloma. Bone Marrow Transplant. 2001;27(10):1037-1043.
doi pubmed - Attal M, Harousseau JL, Facon T, Guilhot F, Doyen C, Fuzibet JG, Monconduit M, et al. Single versus double autologous stem-cell transplantation for multiple myeloma. N Engl J Med. 2003;349(26):2495-2502.
doi pubmed - D'Agostino M, Bertamini L, Oliva S, Boccadoro M, Gay F. Pursuing a curative approach in multiple myeloma: a review of new therapeutic strategies. Cancers (Basel). 2019;11(12).
doi pubmed - Roussel M, Hebraud B, Hulin C, Perrot A, Caillot D, Stoppa AM, Macro M, et al. Health-related quality of life results from the IFM 2009 trial: treatment with lenalidomide, bortezomib, and dexamethasone in transplant-eligible patients with newly diagnosed multiple myeloma. Leuk Lymphoma. 2020;61(6):1323-1333.
doi pubmed - Alexanian R, Dimopoulos M. The treatment of multiple myeloma. N Engl J Med. 1994;330(7):484-489.
doi pubmed - Raje NS, Anaissie E, Kumar SK, Lonial S, Martin T, Gertz MA, Krishnan A, et al. Consensus guidelines and recommendations for infection prevention in multiple myeloma: a report from the International Myeloma Working Group. Lancet Haematol. 2022;9(2):e143-e161.
doi pubmed - Drayson MT, Bowcock S, Planche T, Iqbal G, Pratt G, Yong K, Wood J, et al. Levofloxacin prophylaxis in patients with newly diagnosed myeloma (TEAMM): a multicentre, double-blind, placebo-controlled, randomised, phase 3 trial. Lancet Oncol. 2019;20(12):1760-1772.
doi pubmed - Vickrey E, Allen S, Mehta J, Singhal S. Acyclovir to prevent reactivation of varicella zoster virus (herpes zoster) in multiple myeloma patients receiving bortezomib therapy. Cancer. 2009;115(1):229-232.
doi pubmed - Girmenia C, Ciceri F, Corradini P, Cuneo A, D'Ancona F, Musto P, Risitano AM, et al. Towards personalized prevention of Herpes zoster infection in patients with hematologic diseases or hematopoietic stem cell transplant recipients: a position paper from an <I>ad hoc</I> Italian expert panel. Haematologica. 2024;109(11):3496-3504.
doi pubmed - White PL, Price JS, Backx M. Therapy and management of pneumocystis jirovecii infection. J Fungi (Basel). 2018;4(4).
doi pubmed - Rosen LS, Gordon D, Kaminski M, Howell A, Belch A, Mackey J, Apffelstaedt J, et al. Zoledronic acid versus pamidronate in the treatment of skeletal metastases in patients with breast cancer or osteolytic lesions of multiple myeloma: a phase III, double-blind, comparative trial. Cancer J. 2001;7(5):377-387.
pubmed - Terpos E, Zamagni E, Lentzsch S, Drake MT, Garcia-Sanz R, Abildgaard N, Ntanasis-Stathopoulos I, et al. Treatment of multiple myeloma-related bone disease: recommendations from the Bone Working Group of the International Myeloma Working Group. Lancet Oncol. 2021;22(3):e119-e130.
doi pubmed - Swarm RA, Abernethy AP, Anghelescu DL, Benedetti C, Buga S, Cleeland C, Deleon-Casasola OA, et al. Adult cancer pain. J Natl Compr Canc Netw. 2013;11(8):992-1022.
doi pubmed - Fallon M, Giusti R, Aielli F, Hoskin P, Rolke R, Sharma M, Ripamonti CI, et al. Management of cancer pain in adult patients: ESMO Clinical Practice Guidelines. Ann Oncol. 2018;29(Suppl 4):iv166-iv191.
doi pubmed - Smith EM, Pang H, Cirrincione C, Fleishman S, Paskett ED, Ahles T, Bressler LR, et al. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA. 2013;309(13):1359-1367.
doi pubmed - VanderWall K, Daniels-Wells TR, Penichet M, Lichtenstein A. Iron in multiple myeloma. Crit Rev Oncog. 2013;18(5):449-461.
doi pubmed - Bohlius J, Bohlke K, Castelli R, Djulbegovic B, Lustberg MB, Martino M, Mountzios G, et al. Management of cancer-associated anemia with erythropoiesis-stimulating agents: ASCO/ASH clinical practice guideline update. Blood Adv. 2019;3(8):1197-1210.
doi pubmed - Bohlius J, Schmidlin K, Brillant C, Schwarzer G, Trelle S, Seidenfeld J, Zwahlen M, et al. Recombinant human erythropoiesis-stimulating agents and mortality in patients with cancer: a meta-analysis of randomised trials. Lancet. 2009;373(9674):1532-1542.
doi pubmed - Bastit L, Vandebroek A, Altintas S, Gaede B, Pinter T, Suto TS, Mossman TW, et al. Randomized, multicenter, controlled trial comparing the efficacy and safety of darbepoetin alpha administered every 3 weeks with or without intravenous iron in patients with chemotherapy-induced anemia. J Clin Oncol. 2008;26(10):1611-1618.
doi pubmed - Carson JL, Stanworth SJ, Guyatt G, Valentine S, Dennis J, Bakhtary S, Cohn CS, et al. Red blood cell transfusion: 2023 AABB international guidelines. JAMA. 2023;330(19):1892-1902.
doi pubmed - Terpos E, Kleber M, Engelhardt M, Zweegman S, Gay F, Kastritis E, van de Donk NW, et al. European Myeloma Network guidelines for the management of multiple myeloma-related complications. Haematologica. 2015;100(10):1254-1266.
doi pubmed - Bonilla-Valentin FJ, Cerra J, Caceres-Perkins W, Alsina M. Case report of IgM multiple myeloma: diagnosing a rare hematologic entity. Cancer Control. 2018;25(1):1073274817744448.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.