| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 15, Number 6, December 2024, pages 871-881

Loss of MK2 Enhances Radiation-Mediated Apoptosis in Bladder Cancer
Deri Morgana ![]() , Kiersten L. Berggrena, b,
Grace Millingtona, Hanna Smitha, Colby Spiessa, Michael
Hixonb, Benjamin L. Woolbrightc, John A. Taylor IIIc, Randall
J. Kimpled, Ronald Chena, Xinglei Shena, Gregory N. Gana,
e
, Kiersten L. Berggrena, b,
Grace Millingtona, Hanna Smitha, Colby Spiessa, Michael
Hixonb, Benjamin L. Woolbrightc, John A. Taylor IIIc, Randall
J. Kimpled, Ronald Chena, Xinglei Shena, Gregory N. Gana,
e
aDepartment of Radiation Oncology, University of Kansas Medical Center, Kansas City,
KS, USA
bThe University of New Mexico, School of Medicine, Albuquerque, NM,
USA
cDepartment of Urology, University of Kansas Medical Center, Kansas City,
KS, USA
dDepartment of Human Oncology, University of Wisconsin, Madison, WI,
USA
eCorresponding Author: Gregory N. Gan, Department of Radiation Oncology,
University of Kansas Medical Center, Kansas City, KS, USA
Manuscript submitted August 13, 2024, accepted October 15, 2024, published online December 11,
2024
Short title: MK2 Is Tumorigenic in Bladder Cancer
doi:
https://doi.org/10.14740/wjon1945
| Abstract | ▴Top |
Background: Bladder cancer patients unable to receive cystectomy or who choose to pursue organ-sparing approach are managed with definitive (chemo)radiotherapy. However, this standard of care has not evolved in decades and disease recurrence and survival outcomes remain poor. Identifying novel therapies to combine with radiotherapy (RT) is therefore paramount to improve overall patient outcomes and survival. One approach is to find cellular mechanisms that can be targeted to increase the radiosensitivity of bladder cancer. The stress-activated kinase directly downstream from p38 mitogen-activated protein kinase (MAPK), mitogen-activated protein kinase activated protein kinase 2 (MAPKAPK2 or MK2), has been shown to enhance cancer-mediated inflammation, mesenchymal gene expression, and in vivo tumor growth. Here we examined the impact that MK2 knockdown (KD) has on bladder cancer cell radiosensitivity.
Methods: We utilized short hairpin RNA (shRNA) KD of MK2 using lentiviral transfection in the bladder cancer cell lines, T24 and HTB9. We compared the growth of KD cells to wild type using colony formation assays, proliferation assays and cell counts to determine differences in cell growth. Apoptosis was examined by annexin-based flow cytometry and western blots. Flow cytometry was also used for cell cycle analysis.
Results: KD clones showed a greater than 90% inhibition of MK2 expression as determined by western blot. Clonogenic assays exhibited an increase in radiosensitivity among the MK2 KD bladder cancer cells. These data were supported with proliferation assays that displayed a greater reduction in cell number following RT in MK2 KD bladder cancer cells. Annexin V binding in bladder cancer cells suggested increased apoptosis in MK2 KD cells. This was confirmed by comparing the amount of cleaved caspase products for the caspases 3 and 8 to scrambled control (SCR), and the release of cytochrome C into the cytosol. Both cell types showed disruptions in the cell cycle but at different points in the cycle.
Conclusion: These results show that MK2 controls irradiation-induced apoptosis in bladder cancer cells.
Keywords: Bladder cancer; MK2; Apoptosis; Radiation; shRNA; Caspase 3
| Introduction | ▴Top |
Bladder cancer is the seventh most frequent cancer in the United States and the ninth most deadly [1], with an estimated 83,000 new cases and over 17,000 deaths predicted in the United States in 2021 [2]. Risk factors for bladder cancer include age, genetics, smoking solvent exposure, contaminated drinking water (chlorine, arsenic), cardiovascular disease and previous cancer treatment [3, 4]. There are two types of bladder cancer, non-muscle invasive bladder cancer (NMIBC) which is a superficial form of cancer that remains in the bladder lining, and muscle invasive bladder cancer (MIBC) that has infiltrated the bladder’s muscle layer. For NMIBC, treatment includes transurethral resection or Bacillus Calmette-Guerin immunotherapy [5, 6]. For MIBC, treatment is focused on either radical cystectomy with or without neoadjuvant or adjuvant chemotherapy [7, 8], or tri-modality therapy (TMT) which combines transurethral tumor resection with chemotherapy and radiotherapy (RT) in cases where radical cystectomy is not appropriate [9]. The use of RT alone or in combination with chemotherapy is done in cases of desired bladder sparing [10].
The p38/mitogen-activated protein kinase (MAPK) pathway is activated by cell stress and has been extensively implicated as a tumor promoter, playing a role in cell migration and invasion, proliferation, and activation of inflammatory and immune signaling [11-13]. When the pathway is activated, p38 forms a complex with mitogen-activated protein kinase activated protein kinase 2 (MAPKAPK2 or MK2) and phosphorylates in the nucleus [13, 14], revealing a nuclear export sequence that causes the complex to travel to the cytoplasm. There, the complex activates downstream targets involved in cell proliferation, deoxyribonucleic acid (DNA) damage resistance, and epithelial-to-mesenchymal transition (EMT) in both the nucleus and the cytoplasm [14, 15]. Recent data postulate MK2 as a sensor of signal intensity that regulates cell fate deciding whether cells will survive based on the loss of MK2 [16].
We have previously shown that there is an upregulation in MK2 phosphorylation and increased cytokine and EMT gene expression following RT in head and neck cancer (HNC) and colorectal cancers [17, 18]. Other studies have shown that p38, MK2, and their downstream targets are involved in bladder cancer migration and invasion [19-21]. Treatment of bladder cancer cell lines HTB5 and HTB9 with a p38 inhibitor decreased cell growth and proliferation, blocked DNA synthesis, increased apoptosis, and decreased invasion of HTB9 cells [20, 21]. Furthermore, Kumar and colleagues showed that kinase-inactive mutant MK2 cell lines had less invasion compared to wild-type (WT) MK2 cell lines [20]. Overexpression of heat shock protein 27 (HSP27), a downstream target of MK2, in bladder cancer cell lines promoted cell invasion in vitro, while knockdown (KD) of HSP27 led to decreased tumor growth in vivo [19]. Therefore, targeting the p38-MK2-HSP27 signaling axis may reduce bladder cancer growth by affecting DNA synthesis, migration, and invasion.
One of the functions of MK2 is to act as a cell cycle checkpoint pathway that arrests the cells in response to DNA damage. It was shown that in the absence of a functional p53 isoform, MK2 serves to arrest the cell cycle in G2/S phase in response to DNA damage via UV irradiation [22, 23]. Deletion of MK2 resulted in increased death with increased caspase 3 cleavage. The mechanism of this was proposed to be through the phosphorylation of cell cycle phosphatases M-phase inducer phosphatase 2 and 3 (CDC25B and CDC25C, respectively). These proteins dephosphorylate cyclin-dependent kinases (CDK) proteins and advance the cell cycle [24] under normal conditions. With the activation of MK2, CDC25 proteins are phosphorylated [22, 23, 25] and subsequently targeted for degradation, thus preventing the cell from experiencing mitotic catastrophe. Later studies showed that MK2 was responsible for cisplatin resistance in p53 null cells and that simultaneous inhibition of MK2 restored cisplatin potency and increased the expression of caspase 3 in response to cisplatin treatment [26]. The discovery that MK2 acts as an additional cell cycle checkpoint that runs in parallel to Chk1 was used to show that simultaneous inhibition of Chk1 and MK2 in BRAF or KRAS tumors from patients caused apoptotic cell death [27]. These data led to experiments examining the synthetic lethality of cancer cells to inhibitors of p53 and MK2 [28]. Recent evidence has further identified the importance of MK2 as a checkpoint effector that may be targeted to enhance the radiosensitivity or tumor cell [29].
The data demonstrated the effectiveness of MK2 blockade as a promotor of cell death after UV treatment was performed on U2OS cells [22]. Follow-up work that utilized cytotoxic agents rather than radiation as a stimulator of DNA damage [23, 25] focused on the same cells or mouse embryonic fibroblasts (MEFs). Therefore, whether the same mechanisms would play a role in other cells is unclear. Non-surgical bladder cancer therapy has largely remained unchanged over multiple decades. Because RT is routinely used in the non-surgical management of this disease, novel therapies combined with RT may enhance local and regional disease control. Based on published data, we hypothesize that genetic KD of bladder cancer cells will sensitize them to RT. Here we show different levels of radiosensitivity in bladder cancer cells with p53 mutant backgrounds [30-32].
| Materials and Methods | ▴Top |
Cell culture
T24 cells and HTB9 cells were obtained from ATCC. Cell lines were verified by short tandem repeat analysis (University of Arizona Genetics Core, Tucson, AZ) and confirmed negative for mycoplasma infection by Plasmotest™ (InvivoGen, San Diego, CA). Cells were maintained in full growth medium (Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin). Cells were kept viable by passaging the cells 1:8 every 3 days. Cells were trypsinized with 0.05% trypsin/ethylenediaminetetraacetic acid (EDTA) for 5 min at 37 °C followed by adding fresh media to neutralize.
For short hairpin RNA (shRNA) KD experiments, MK2 expression was reduced in T24 and HTB9 cells using pGFP-C-Lenti shRNA lentiviral plasmids targeted to MAPKAPK2 (Origene, Rockville, MD). Cells were transfected with scramble or shRNA constructs and transfected cells were selected by treatment with puromycin (2 µg/mL for T24; 0.5 µg/mL for HTB9), isolated, and maintained under standard culture conditions. Transfection was verified by positive green fluorescent protein (GFP) signals in cells in conjunction with the addition of puromycin for continued selection. GFP positive and puromycin resistant cells were clonally isolated and were selected for use based on the expression of MK2 by immunoblot. Scramble and shRNA transfected T24 and HTB9 were treated with or without 10 Gy radiation, harvested at various time points, and lysates were processed for protein and mRNA analysis.
Protein analysis
For protein analysis, cells were grown in 90 mm dishes so that they were in log-phase growth (79-90% confluent) at the time of lysis. Media was rapidly removed from dishes, plates were washed with ice-cold phosphate buffer saline (PBS) and cells were lysed using a cell scraper and 100 - 150 µL of radioimmunoprecipitation assay (RIPA) buffer (ThermoFisher, Waltham, MA) containing HALT™ proteinase and phosphatase inhibitors (ThermoFisher, Waltham, MA). Cells were incubated on ice for 10 min and centrifuged at 21,000 g for 10 min. The supernatant was collected and protein concentrations were measured using a standard bicinchoninic acid (BCA) protein assay kit (ThermoFisher, Waltham, MA), followed by western blot analysis by loading 20 - 40 µg of protein onto NuPAGE 4-12% bis-tris gels (ThermoFisher, Waltham, MA) run in the XCell SureLock mini and midi western blot apparatus (ThermoFisher, Waltham, MA). Molecular mass of proteins was estimated by using Novex Sharp pre-stained protein standard (ThermoFisher, Waltham, MA). Gels were transferred onto polyvinylidene difluoride (PVDF) membranes using the iBlot 2 dry transfer apparatus (ThermoFisher, Waltham, MA) and desired protein bands were identified by overnight incubation of primary antibodies at 4 °C in tris-buffered saline with Tween 20 (TBST) buffer with either 5% milk (Lab Scientific, Danvers, MA) or 3% bovine serum albumin (BSA) (Gemini Bioscience, West Sacramento, CA). Desired protein bands were identified by incubating the membranes with anti-rabbit or mouse IgG horseradish peroxidase (HRP) linked antibodies for 30 min at room temperature before using SuperSignal™ West Femto (ThermoFisher, Waltham, MA) to react with the antibody bound HRP. Membranes were visualized on CL-Xposure™ film (ThermoFisher, Waltham, MA). For cell fractionation experiments, we utilized the cell fractionation kit (Cell Signaling, Danvers, MA).
Antibodies for western blot analysis were obtained from Cell Signaling: MK2, p-MK2, HSP27, p-HSP27, p38, p-p38, vinculin, caspase3, cleaved caspase 3, caspase 6, cleaved caspase 6, caspase 7, cleaved caspase 7, caspase 8, cleaved caspase 8, CDC25b, cytochrome C, and poly-ADP ribose polymerase (PARP); ThermoFisher: p-MK2 (S272), p-CDC25B.
Irradiation
Cells were irradiated in different growing vessels depending on the assay. CyQUANT assays utilized 96-well plates; colony forming analysis, annexin binding and cell cycle were performed in six-well dishes. The growth container containing live cells and media were placed on a raised turntable inside a J.L. Shepherd Mark I 68-A cesium irradiator (J.L. Shepherd & Associates, San Fernando, CA). The turntable was spun at 6 rpm for the calculated time as determined by a calculation to correct for element decay. Immediately after irradiation, the growth media was replaced with fresh warm media and the cells were left to incubate until time of harvest.
Quantitative PCR
For mRNA analysis, RNA was extracted from cell lysates using a Qiagen RNeasy kit following the manufacturer’s instructions (Qiagen, Hilden, Germany) and mRNA was quantified on a nanodrop (Cell Signaling, Danvers, MA). cDNA was synthesized using a Verso cDNA synthesis kit (ThermoFisher) and samples were analyzed by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) on a CFX Thermal Cycler (BioRad, Hercules, CA). Probes for RT-qPCR analysis were obtained from ThermoFisher (Waltham, MA). FxcycleTMPI/RNAase staining solution was used for cell cycle analysis and Annexin V 647 conjugate for annexin binding experiments was obtained from ThermoFisher (Waltham, MA).
Quantitation of the PCR reactions was performed using the ΔΔCT method where the gene target CT counts were normalized to the CT counts of either ubiquitin or GAPDH which were used as endogenous reference genes.
Proliferation assays
Cells were harvested by trypsinization during logarithmic growth and reseeded into 96-well plates (Fisher Scientific) at 5 - 10 × 104 cells/well in 200 µL of complete growth media plus the required puromycin concentration. The cells were allowed to attach to the plate for 8 h at which point the first time point was collected for analysis. At the time points specified growth media was removed from the culture plates by upending in a sink, washed with cold PBS and frozen dry at -80 °C until analysis. Relative cell growth was measured using CyQUANT™ cell proliferation assay (Cell Signaling, Danvers, MA) on an Infinite M200Pro (Tecan, Mannedorf, Switzerland) ex. 480, em. 420. Cell growth is expressed as a percentage of fluorescence at time 0.
Annexin binding apoptosis assays
Cells were plated in 60 mm petri dishes at 0.15 - 0.6 × 106 cells/dish to ensure that the cell lines were in logarithmic growth at the time of harvest. Growth media was removed and placed in separate FACS tubes and placed on ice. The dishes were washed twice with warm PBS, then warm 0.05% trypsin was added to the cells. Cells were coaxed off rapidly with trituration and the cell suspension was added to the cold media in its respective tubes. Cells were centrifuged at 400 g for 5 min, the growth media was removed, and cells were washed twice with cold PBS. The cell pellet was resuspended in 100 µL of annexin binding buffer, with 0.1 µg/mL annexin V conjugated to Alexa Fluor 647 (ThermoFisher, Waltham, MA), and 0.1 µg/mL propidium iodide (PI) then incubated in the dark for 15 min at room temperature. Cell cycle analysis was performed on an LSR II (BD Biosciences, Franklin Lakes, NJ). The intensity of cell fluorescence was plotted on the x-axis with number of cells on the y-axis. This plot resulted in two peaks with a plateau between them. The first peak was quantified as G0/G1 phase, the plateau was considered S phase and the second peak was taken as G2M. The number of cells in the different compartments was transformed into a percentage of total cells analyzed by the flow cytometer. The flow cytometry data were analyzed by FlowJo (FlowJo Software).
Cell cycle analysis
After irradiation, cells were incubated until the chosen time point, trypsinized and pelleted at 400 g at 4 °C. The cell pellet was resuspended in 0.5 mL of cold PBS and added to 4.5 mL of ice-cold 70% ethanol (EtOH). Samples in EtOH were kept at -20 °C until analysis. At the time of analysis, the cells were rehydrated by resuspending in cold PBS for 60 s, centrifuged at 400 g for 5 min and resuspended in 1 mL PI/Triton X-100 staining solution with RNase A. The cell suspension was incubated at room temperature in the dark for 10 min and analyzed on an LSR II (BD Biosciences, Franklin Lakes, NJ). Gating was based on unstained cells compared to untreated cells and cells that had been exposed to 60 °C for 20 min. Cells positive for annexin but without staining for PI were considered early apoptosis. Cells with both high PI and annexin staining were considered late apoptosis and cells only expressing PI fluorescence were considered necrotic. The data were analyzed in FlowJo (FlowJo Software).
Statistical analysis
For all experiments, data handling and statistical analysis were carried out using GraphPad Prism 9 software. One-way analysis of variance (ANOVA) analysis and appropriate post-hoc comparisons were used.
| Results | ▴Top |
Characterization of bladder cancer cell MK2 KD by western blot
To investigate the hypothesis that MK2 alters radiosensitivity of bladder cancer cells, we needed to examine what the specific KD of MK2 would do to the growth and survival of bladder cancer cells by knocking down MK2 expression in two bladder cancer cell lines (T24 and HTB9) using shRNA constructs and comparing their function to a shRNA scrambled control (SCR) (Fig. 1a, b). The shRNA treatment was shown to reduce the expression of MK2 by greater than 90%, as determined by densitometry in blots where the MK2 band was visible in the KD cell line. We used western blot to analyze the phosphorylation states of p38, MK2 and HSP27 at 2, 24 and 48 h after irradiation. These time points were selected to observe both the early responses and the longer-term effects of this pathway activation in response to radiation, which we have performed previously in HNC cells [17].
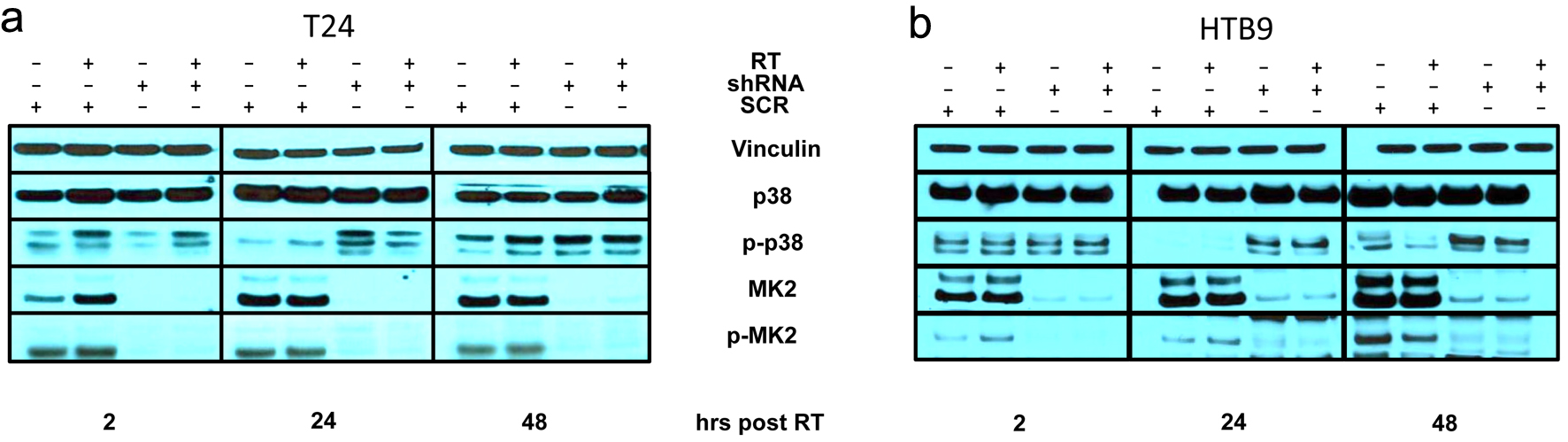 Click for large image |
Figure 1. Immunoblot of SCR or MK2 shRNA of (a) T24 and (b) HTB9 with and without 10 Gy radiation at 2, 24 and 48 h for p38, MK2 and HSP27 with their respective phosphoproteins and vinculin as a loading control. SCR: scrambled control; MK2: mitogen-activated protein kinase activated protein kinase 2; shRNA: short hairpin RNA; HSP27: heat shock protein 27. |
We saw increased phosphorylation of p38 (p-p38) and increased phosphorylation of MK2 at site T334 (p-MK2 T334) shortly after RT compared to non-irradiated controls (Fig. 1a). In the MK2 KD samples, we saw an increase in p-p38 at 24 h in the absence of MK2, demonstrating that there is a disruption of the p38-MK2 signaling axis. Furthermore, we observed that p38 remained phosphorylated in our MK2 KD bladder cancer cell after 48 h. The increased p-p38 at 24 and 48 h indicates that decreasing MK2 protein produces a lasting effect on the p38 pathway after irradiation. The bladder cancer cells were discordant regarding the phosphorylation of HSP27 (Supplementary Material 1A, B, wjon.elmerpub.com). Whereas MK2 KD reduced HSP27 phosphorylation in T24 at all time points shown, HTB9 KD cells showed an increase in phosphorylation at 24 and 48 h which was absent in the WT cells.
Analysis of the effect of MK2 KD on bladder cancer radiosensitivity by clonogenic survival
In agreement with our hypothesis, loss of MK2 led to increased radiosensitivity in bladder cancer in a dose-dependent fashion (0 - 10 Gy) in traditional clonogenic survival assays. HTB9 and T24 MK2 KD cells were significantly more sensitive to radiation compared to SCR cells, with the significance in sensitivity beginning at 6 Gy for HTB9 cells (surviving fraction at 6 Gy = 0.216 ± 0.04 in SCR compared to 0.06 ± 0.013 in shRNA; P = 0.0102) and at 4 Gy for T24 (surviving fraction at 4 Gy = 0.234 ± 0.014 in SCR compared to 0.089 ± 0.004 in shRNA cells; P = 0.0003) (Fig. 2a, b).
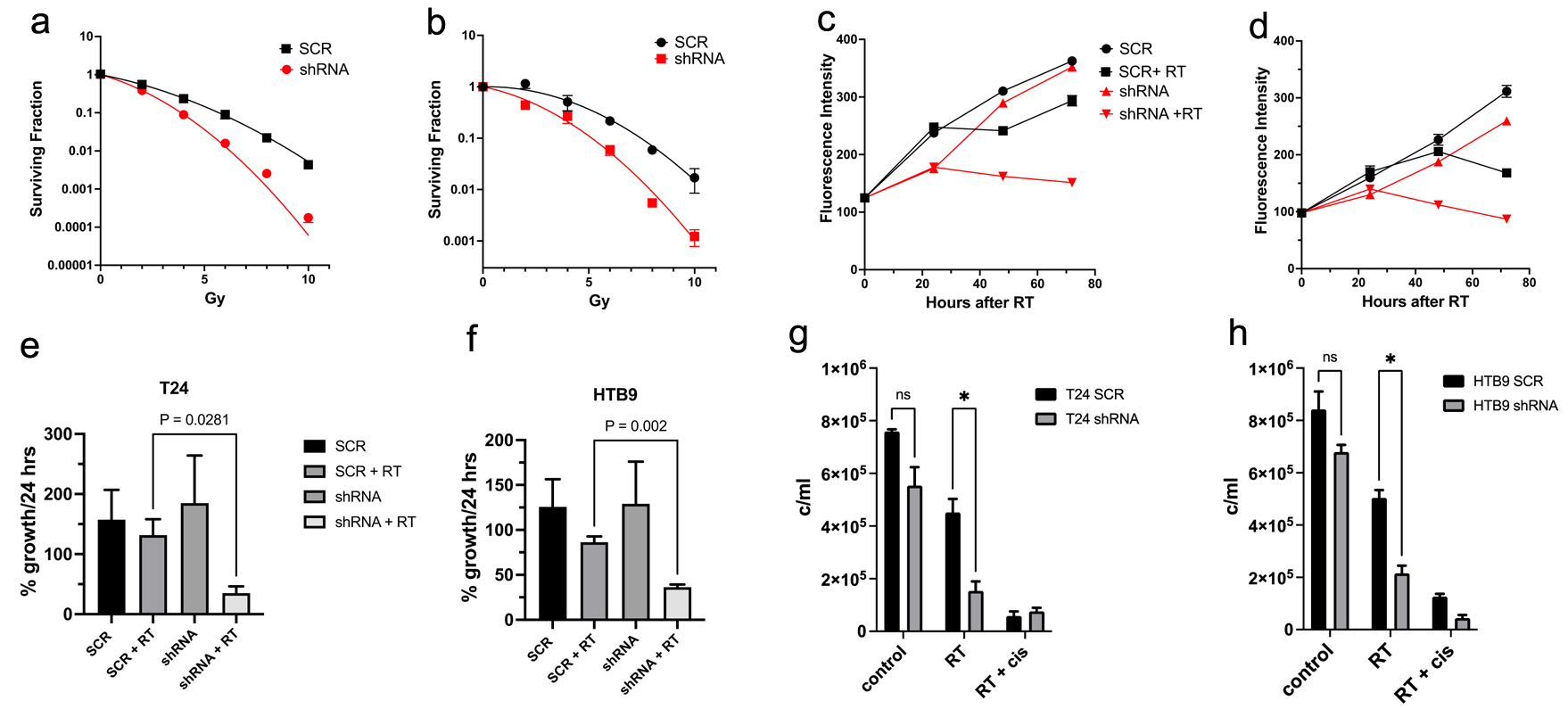 Click for large image |
Figure 2. Clonogenic survival curves of SCR vs. shRNA for the cell lines of (a) T24 and (b) HTB9 cells treated with increasing doses of radiation from 2 to 10 Gy. The data are mean ± SEM, N = 3. Proliferation measurements comparing SCR vs. MK2 shRNA for 72 h of (c) T24 and (d) HTB9 cells with and without 10 Gy radiation. Data are representative of three experiments performed in triplicate. Maximum rate of growth in 24 h expressed as percentage of the maximum growth in SCR cells without RT in (e) T24 and (f) HTB9 cells. Data are mean ± SEM, n = 3. Live cell counts of cells by trypan blue exclusion of (g) T24 SCR vs. shRNA and (h) HTB9 SCR vs. shRNA after 48 h of 10 Gy RT. For a positive control, 25 mM cisplatin + 10 Gy RT was used. SCR: scrambled control; MK2: mitogen-activated protein kinase activated protein kinase 2; shRNA: short hairpin RNA; RT: radiotherapy; SEM: standard error of the mean. |
The effect of MK2 KD and radiation on bladder cancer cell proliferation
The hypothesis that MK2 KD increases radiosensitivity in bladder cancer cells was also supported by proliferation assays measuring the quantity of DNA in a cell culture. For the pattern of cell proliferation in the MK2 KD cells compared to SCR (Fig. 2c, d), SCR cells showed more resilience to RT with a small delay in proliferation compared to the more prolonged dying of MK2 KD cells, where growth rates were negative for 48 h post RT. Growth rates of SCR compared to MK2 KD cells showed similar patterns when comparing maximum rate of growth over the course of 72 h. T24 SCR reached a growth rate of 157±49%/24 h and was similar to 185±79%/24 h in T24 KD cells. After radiation T24 SCR cells reached a maximum rate of 131±7%/24 h compared to 35 ± 47.2 h for irradiated T24 KD cells (P = 0.028) (Fig. 2e). Similarly, HTB9 SCR cells showed a maximum growth rate of 125±30%/24 h compared to 129±46% in the KD cells. After radiation HTB9 SCR had a maximum growth of 86±6.5%/24 h compared to 36 ±2%/24 h in the MK2 KD group (P = 0.002) (Fig. 2f).
Analysis of the effect of MK2 KD and radiation on bladder cancer cell viability
The increased radiosensitivity of the KD cells should decrease the viability of KD compared to WT if our hypothesis is to be valid. Therefore, we examined the trypan blue exclusion of KD cells compared to SCR. Figure 2g (T24) and h (HTB9) shows the differences in viable cell counts 48 h after radiation as determined by trypan blue exclusion. There were significantly fewer cells left in the KD groups compared to the SCR groups after 48 h (T24 SCR 4.5 ± 0.2 × 105 cells vs. T24 KD 1.5 ± 2 × 105; HTB9 SCR 5.0 ± 105 cells vs. HTB9 KD 2.2 ± 0.3 × 105 cells) in agreement with our hypothesis. Cisplatin at a concentration of 25 µM was used in conjunction with 10 Gy radiation as a positive control.
Analysis of MK2 KD and radiation on cancer cell apoptosis by annexin binding and PI staining
Due to the observed increase in bladder cancer radiosensitivity in MK2 KD cell lines, we hypothesized that the increase in radiosensitivity was due to an increase in apoptosis in the cancer cells. We analyzed sets of SCR and KD cells with and without RT for expression of annexin and PI permeability to determine any increase in apoptosis in shRNA cells compared to SCR (Fig. 3). In both cell lines, there was a greater loss of viable cells in KD cells compared to SCR cells at both 24 and 48 h following RT (Fig. 3a, b, e: T24 SCR 56±1.1% at 24 h following RT and 49.8±12.3% at 48 h compared to T24 shRNA 29.9±3.4% at 24 h and 20.1±8.2% at 48 h post RT; Fig. 3c, d, f: HTB9 SCR 68.1 ± 1.9 at 24 h and 45.2 ± 4.7 at 48 h compared to HTB9 shRNA 45.5 ± 1.6 at 24 h and 29.6 ± 6.2 at 48 h; P < 0.05 for all comparisons). The annexin binding/PI staining assay showed no increase in necrosis following RT, which was defined by cells permeable to PI, with no increase in annexin staining. However, when 10 µM cisplatin was added as a positive control, there was a marked increase in necrosis in both cell types at 24 and 48 h. HTB9 cells showed a significant increase in cells undergoing early apoptosis at 24 h post RT and a significant increase in late apoptosis 48 h post RT (Fig. 3c, d, f: HTB9 early apoptosis: 24 h SCR 17±1.2% compared to KD 34.7±1.4%, P < 0.0001; HTB9 late apoptosis: 48 h SCR 29.7±7.1% compared to KD 45.5±8.9%, P = 0.032). T24 cells showed a consistent and significant increase in the percentage of cells that were in late apoptosis at both 24 and 48 h with no significant increase in early apoptotic cells at either 24 or 48 h (T24 late apoptosis: 24 h SCR 20.5±3.9% compared to KD 32.0±5.0%, P = 0.033; T24 late apoptosis: 48 h SCR 18.4±5.6% compared to shRNA 35.9±7.1%, P = 0.0085).
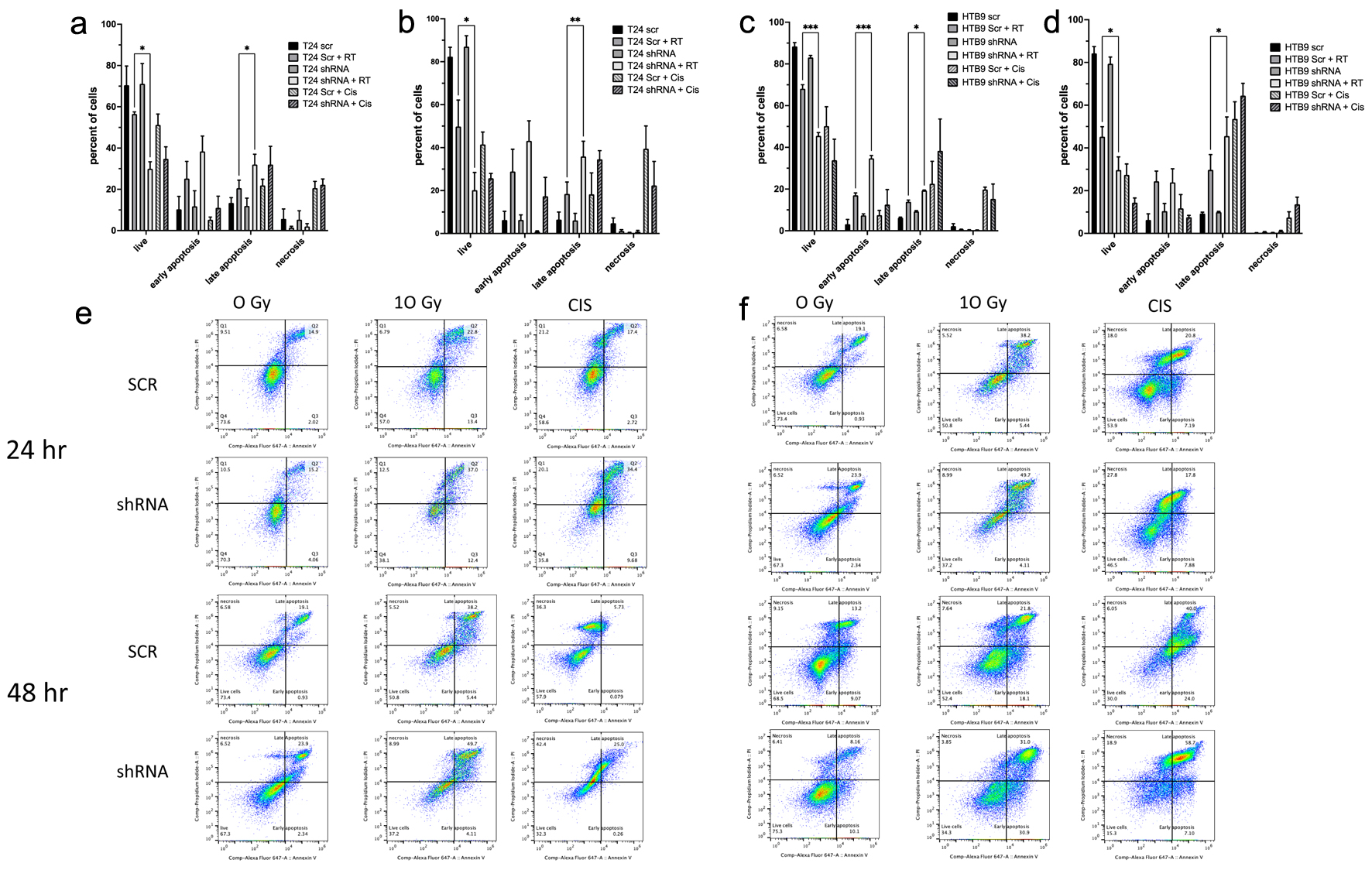 Click for large image |
Figure 3. The analysis of apoptosis by annexin binding and flow cytometery. Data showing T24 SCR vs. shRNA cells at (a) 24 h and (b) 48 h post 10 Gy RT are shown in a histogram (data are mean ± SEM, n = 3) and are also as dot plots (e) from representative experiments. HTB9 SCR vs. shRNA cells show (c) 24 and (d) 48 h after RT in both histogram and dot blots (f) in a similar fashion. For a positive control, 10 mM cisplatin was used in both sets of data. All data are shown as mean ± SEM, n = 3. SCR: scrambled control; shRNA: short hairpin RNA; RT: radiotherapy; SEM: standard error of the mean. |
Measurement of the effect of MK2 KD and radiation on bladder cancer cell apoptosis by western blot
To further examine the hypothesis that the increase in radiosensitivity in MK2 KD cells results in an increase in apoptosis in KD cells shown by the changes of annexin binding and PI staining, we looked for apoptosis markers in cell lysates of HTB9 and T24 cells with and without RT. We looked at samples 12 or 24 h post RT and analyzed the samples for the presence of cleaved caspase and PARP products via immunoblot. In both cell types, there was a greater amount of cleaved caspase 3, 8 and PARP both 12 and 24 h following RT in KD cells compared to SCR cells (Fig. 4a, b). T24 KD cells showed an increase in cleaved caspase 6 at 12 h compared to SCR, which was gone at 24 h, Whereas HTB9 KD showed an increase in cleaved caspase 6 compared to SCR at 24 h but not at 12 h.
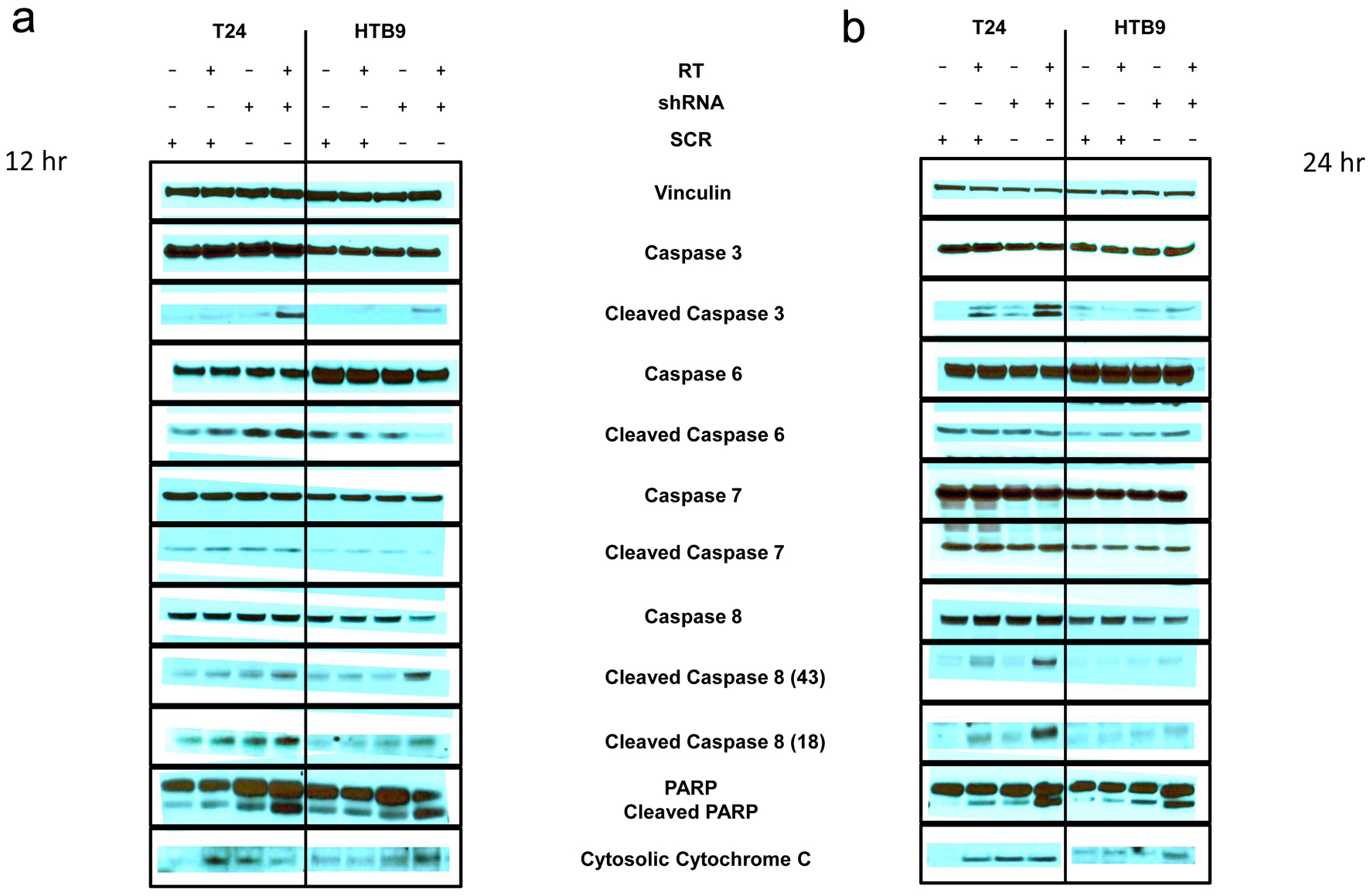 Click for large image |
Figure 4. Immunoblot of apoptosis markers (a) 12 h and (b) 24 h after radiation in T24 SCR vs. shRNA with and without 10 Gy RT (left panels) and HTB9 SCR vs. shRNA with and without RT (right panels). Vinculin is used as a loading control. SCR: scrambled control; shRNA: short hairpin RNA; RT: radiotherapy. |
The effect of MK2 KD and radiation on bladder cancer cell cycle patterns
We hypothesized that the increased apoptosis of MK2 KD would show a perturbation of cell cycle response after irradiation. We stained cells with PI and examined the different cell cycle peaks (G0, G1, S, G2M) to determine if MK2 KD altered the cell cycle profile after radiation. Figure 5 shows the cell cycle profiles of HTB9 SCR, HTB9 KD (Fig. 5c) and T24 SCR, T24 KD (Fig. 5a) over 48 h after 10 Gy irradiation. The different panels show a consistent pattern, where irradiation causes a precipitous drop in the number of cells in G1 (50-60%) to about 10% at 18 h following RT. This is followed by a recovery to 15-20% by 42 h. The biggest difference between the time course of the respective SCR vs. KD occurred at 24 h following RT and thus we chose this time point to examine the cell cycle changes in more detail. Figure 5b, d shows several graphs that compare the percentage of cells in the different cell cycle positions 24 h after RT.
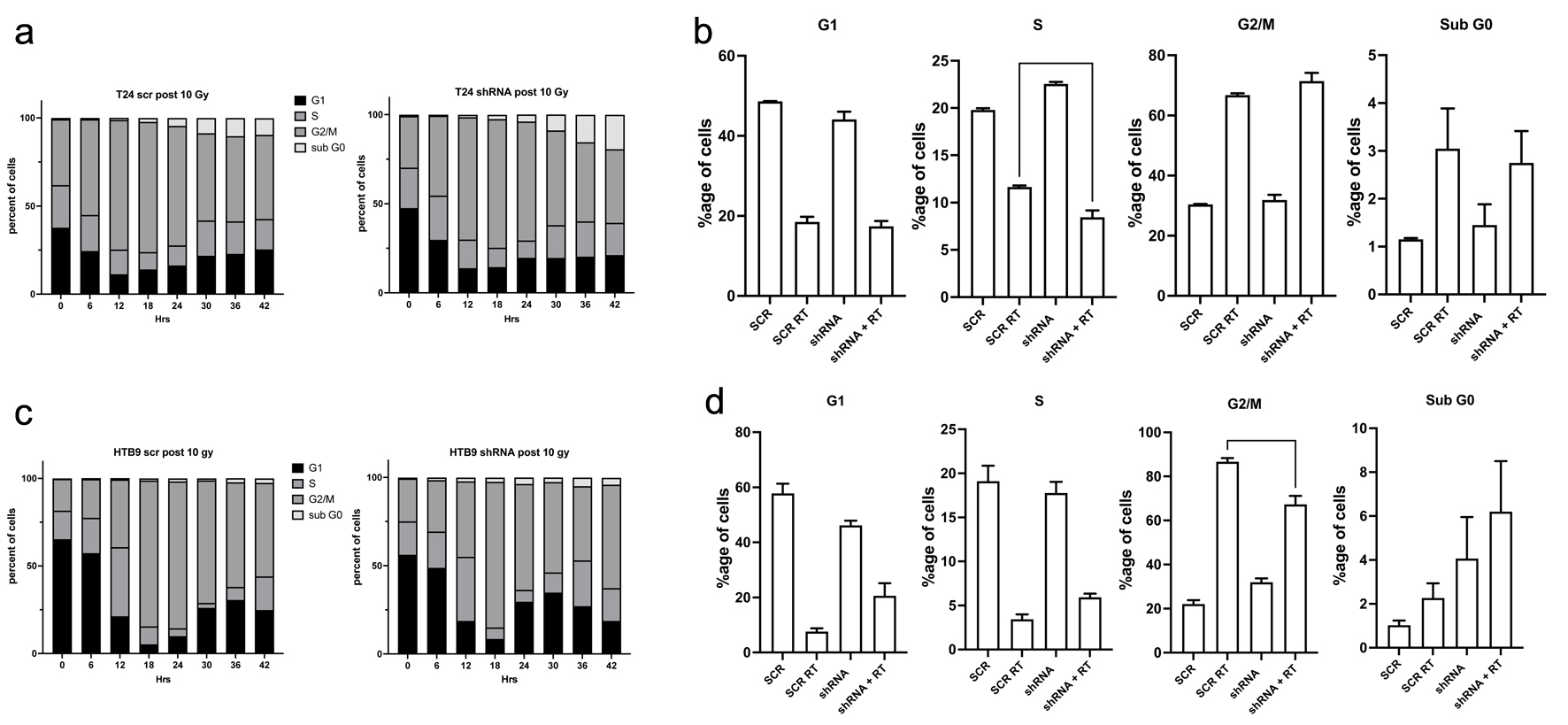 Click for large image |
Figure 5. Cell cycle analysis of T24 SCR vs. shRNA cells (a, b) and HTB9 SCR vs. shRNA cells (c, d) as determined by propidium iodine (PI) staining post 10 Gy RT. a and c show a representative time course of the changes in cell cycle progression after a dose of 10 Gy irradiation to (a) T24 SCR vs. shRNA cells and (b) HTB9 SCR vs. shRNA cells. c and d show histograms of the differences in the percentage population of cells in the various cell cycle stages 24 h after 10 Gy RT (mean ± SEM, n = 3). SCR: scrambled control; shRNA: short hairpin RNA; RT: radiotherapy; SEM: standard error of the mean. |
HTB9 KD cells exposed to RT showed a smaller number of cells in the G2/M checkpoint compared to HTB9 SCR cells (KD HTB9: 67.3±3.8% compared to 86.7±1.7% in HTB9 SCR; P = 0.0022). This difference was not seen between the T24 SCR and T24 KD cells but there was a greater number of T24 SCR cells in S phase compared to T24 shRNA (8.4 ± 0.7 in T24 KD cells compared to 11.7 ± 0.1 in T24 SCR cells). The in-depth cell cycle analysis of the bladder cancer cells did not elucidate a clear, common mechanism shared between the T24s and HTB9 cell lines that would clarify the observed apoptosis increase.
| Discussion | ▴Top |
We explored the hypothesis that KD of MK2 in bladder cancer showed an increase in radiotherapy-mediated cell death and found support for the hypothesis as measured by colony forming assays, proliferation assays, and annexin binding assays. We also show that in conjunction with the increased cell death, there was a greater expression of cleaved PARP, cytosolic cytochrome C, and caspase products, consistent with an increase in apoptosis and our hypothesis. To our knowledge, this is the first characterization of the effect of RT on MK2 pathway activation and the impact of MK2 inhibition on enhancing tumor radiosensitivity leading to enhanced apoptosis in bladder cancer cell lines.
Previous work has shown that MK2 acts as an inhibitor of cell cycle progression after DNA damage in cells that have mutated p53 [22, 23]. These studies demonstrated that CDC25b is a phosphorylation target for MK2 [22, 33]. CDC25b is a member of the CDC25 family of phosphatases that are involved in a mechanism where the cell cycle regulators, Chk1 and Chk2, control the DNA damage response. CDC25 phosphatases positively regulate cyclins, pushing the cell cycle forward. In response to DNA damage, these proteins are phosphorylated by MK2, causing them to bind to 14-3-3 proteins that sequester the molecule away from the nuclear site and into the cytosol [24]. The result is cell cycle arrest at the G2/M checkpoint that serves as a tertiary checkpoint in the absence of a functional p53. The studies examining the pharmacological or genetic modification of MK2 activity showed that DNA damage caused by UV or genotoxic stress resulted in increased cell death due to mitotic catastrophe in MEFs [22]. We also saw a change in the cell cycle after radiation in both sets of bladder cancer cells, but the change was much smaller than what was shown in the U2SO cells. In response to radiation, there was a reduction in the number of cells in G1 phase to roughly 10% G1 and 80% G2/M at 18 - 24 h. This was seen in both KD and SCR cell lines. HTB9 shRNA cells showed a 20% reduction of cells in the G2/M phase compared to SCR cells at 24 h, which was significant but much lower than the 45% change seen in the previous work on U2O2 cells. The T24 KD cells failed to show any significant change compared to SCR regarding the number of cells in the G2/M phase 24 h after RT. However, there was a slight but significant reduction of cells in the S phase.
The difference between the expected changes in the cell cycle after undergoing irradiation (based on the Manke et al’s study [22]) and the observation that CDC25b shows few phosphorylation changes between SCR and KD (Supplementary Material 1, wjon.elmerpub.com) suggests that CDC25b was not a primary driver of cell death in these cells. Whereas there was a reduction of CDC25B phosphorylation at 2 h in the T24 MK2 KD cells compared to SCR, this reduction was absent by 24 h when the greatest differences in cell cycle populations were seen. In the HTB9 cells, there was no change in the phosphorylation status of CDC25b at any of the time points tested, even though MK2 phosphorylation is increased at the 2 h mark (Fig. 1b). We therefore suggest that while the presence of MK2 affects the phosphorylation state of CDC25b, it may not play a significant role in cell cycle arrest in these cells. Our findings challenge the traditional notion that MK2-mediated regulation of CDC25b is necessary for apoptosis in bladder cancer. Loss (or inhibition) of MK2 appears to enhance RT-mediated apoptosis in a CDC25b independent fashion.
When we profiled the caspase products after irradiation, we found a greater expression of cleaved caspase 3, and 8 (Fig. 4). The mechanism is not clear, by which depletion of MK2 in bladder cells showed greater apoptosis after irradiation. These cells have mutant p53 [30-32] and therefore MK2 should serve as a secondary checkpoint. MK2 inhibition is known to inhibit the release of tumor necrosis factor-alpha (TNF-α) and other cytokines (interleukin (IL)-6, interferon-gamma (IFN-γ), IL-1α) [17, 18, 34-36]. This inhibition is dependent on tristetraprolin (TTP), which normally gathers the mRNA for these cytokines and degrades them, thus reducing the transcription of the message [37]. Phosphorylation of TTP by MK2 prevents TTP from binding to these inflammatory cytokine mRNA species and marking them for degradation. TTP activation has been shown to increase the apoptosis of glioma cells by downregulating urokinase plasminogen activator [38, 39]. Therefore, it is possible that the dysregulation of MK2 in the bladder cancer cells caused an increase in activated TTP after radiation, resulting in a greater level of apoptosis.
Another potential mechanism for modulating apoptosis is the direct downstream target of MK2, HSP27. It was one of the first substrates of MK2 discovered, and it has a wide range of activities in cell survival, including the regulation of apoptosis through its interaction with cytochrome C [40]. Our supplementary data show the modulation of HSP27 phosphorylation after the KD of MK2 is changed in T24 shRNA cells compared to control cells, and this could alter the sensitivity of cells to cytochrome C release. However, the lack of significant changes to HSP27 phosphorylation in HTB9 cells suggests that the effects on apoptosis are separate to the effects of MK2 on the phosphorylation of HSP27.
Recent data have implicated MK2 in the cytosolic transport of caspase 3 initiating apoptosis in lung cancer cells [41, 42] with higher levels of MK2 defining positive treatment response. Our data stand in contrast to this paper and other manuscripts referenced within that demonstrate a survival benefit to MK2 by acting as a cell cycle checkpoint. It is possible that similar mechanisms exist in bladder cancer cells; however, the cleavage of caspase 3 in this study was perhaps the strongest indicator of apoptosis in these cells which suggests that different mechanisms play a role in bladder cancer cells compared to lung cancer. Enhancing the argument for tissue specific responses to MK2, MK2 levels were shown to be predictive of poor outcome in pancreatic ductal carcinoma (PDAC), inhibition of MK2 decreased PDAC development, progression and metastasis [43]. In the PDAC study blockade of MK2 resulted in blockage of HSP27 phosphorylation and the prevention of Beclin 1 from promoting protective autophagy pushing the cells towards apoptosis. The authors suggest in the manuscript that PDAC apoptosis was caused by autocrine effects of TNF-α, TNFR1 and TAK1-P38 signaling [43]. We cannot discount that these mechanisms contribute to the increased radiosensitivity of the bladder cancer cells.
Previous studies examining MK2 KD in bladder cancer cells mainly focused on the changes in migration and the activity of MMP2 and MMP9, which are reduced in MK2 KD cells [20]. Here we tested the hypothesis that MK2 affects bladder cancer cell radiosensitivity and show for the first time that MK2 KD sensitizes bladder cancer cells to radiation by enhancing cancer cell apoptosis. Given the dearth of novel targetable therapies for bladder cancer, additional studies will be necessary to determine whether MK2 inhibition combined with RT can enhance disease control and improve overall survival in bladder cancer patients.
| Supplementary Material | ▴Top |
Suppl 1. Immunoblot of (A) T24 and (B) HTB9 SCR vs. shRNA with and without 10 Gy RT after 2, 24 and 48 h.
{kind=link}
Acknowledgments
We acknowledge the Flow Cytometry Core Laboratory, which is sponsored, in part, by the NIH/NIGMS COBRE grant (P30GM103326).
Financial Disclosure
This work was supported by the Kansas Institute for Precision Medicine NIGMS COBRE grant (P20GM130423) and Cancer Center Support Grant (P30CA168524).
Conflict of Interest
None to declare.
Informed Consent
Not applicable.
Author Contributions
D. Morgan: study conception and design, data collection, analysis, interpretation, manuscript writing and preparation; K.L. Berggren: data collection, analysis and manuscript preparation; G. Millington, H. Smith and C. Spiess: data collection; M. Hixon and J.A. Taylor III: manuscript review and preparation; B.L. Woolbright, R.J. Kimple, R. Chen and X. Shen: interpretation, manuscript review and preparation; G.N. Gan: study conception and design, interpretation, manuscript writing, editing and preparation.
Data Availability
All data generated and analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
DNA: deoxyribonucleic acid; EMT: epithelial-to-mesenchymal transition; HSP27: heat shock protein 27; KD: knockdown; MAPK: mitogen-activated protein kinase; MEFs: mouse embryonic fibroblasts; MIBC: muscle invasive bladder cancer; MK2 or MAPKAPK2: mitogen-activated protein kinase activated protein kinase 2; PARP: poly-ADP ribose polymerase; RT: radiotherapy; SCR: scrambled control; shRNA: short hairpin ribonucleic acid; TMT: tri-modality therapy; TTP: tristetraprolin
| References | ▴Top |
- Halaseh SA, Halaseh S, Alali Y, Ashour ME, Alharayzah MJ. A
review of the etiology and epidemiology of bladder cancer: all you need to know. Cureus.
2022;14(7):e27330.
doi pubmed - Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics,
2021. CA Cancer J Clin. 2021;71(1):7-33.
doi pubmed - Alouini S. Risk factors associated with urothelial bladder
cancer. Int J Environ Res Public Health. 2024;21(7).
doi pubmed - Barone B, Finati M, Cinelli F, Fanelli A, Del Giudice F, De
Berardinis E, Sciarra A, et al. Bladder cancer and risk factors: data from a multi-institutional
long-term analysis on cardiovascular disease and cancer incidence. J Pers Med.
2023;13(3).
doi pubmed - Ferro M, Tataru OS, Musi G, Lucarelli G, Abu Farhan AR,
Cantiello F, Damiano R, et al. Modified Glasgow prognostic score as a predictor of recurrence in
patients with high grade non-muscle invasive bladder cancer undergoing intravesical bacillus
Calmette-Guerin immunotherapy. Diagnostics (Basel). 2022;12(3).
doi pubmed - McNall S, Hooper K, Sullivan T, Rieger-Christ K, Clements M.
Treatment modalities for non-muscle invasive bladder cancer: an updated review. Cancers (Basel).
2024;16(10).
doi pubmed - Lenis AT, Lec PM, Chamie K, Mshs MD. Bladder cancer: a
review. JAMA. 2020;324(19):1980-1991.
doi pubmed - Lobo N, Mount C, Omar K, Nair R, Thurairaja R, Khan MS.
Landmarks in the treatment of muscle-invasive bladder cancer. Nat Rev Urol.
2017;14(9):565-574.
doi pubmed - Vashistha V, Wang H, Mazzone A, Liss MA, Svatek RS,
Schleicher M, Kaushik D. Radical cystectomy compared to combined modality treatment for
muscle-invasive bladder cancer: a systematic review and meta-analysis.
Int J Radiat Oncol Biol Phys. 2017;97(5):1002-1020.
doi pubmed - Patel VG, Oh WK, Galsky MD. Treatment of muscle-invasive and
advanced bladder cancer in 2020. CA Cancer J Clin. 2020;70(5):404-423.
doi pubmed - Roux PP, Blenis J. ERK and p38 MAPK-activated protein
kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev.
2004;68(2):320-344.
doi pubmed - Martinez-Limon A, Joaquin M, Caballero M, Posas F, de Nadal
E. The p38 pathway: from biology to cancer therapy. Int J Mol Sci. 2020;21(6).
doi pubmed - Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling
pathways in cancer. Oncogene. 2007;26(22):3279-3290.
doi pubmed - Morgan D, Berggren KL, Spiess CD, Smith HM, Tejwani A, Weir
SJ, Lominska CE, et al. Mitogen-activated protein kinase-activated protein kinase-2 (MK2) and
its role in cell survival, inflammatory signaling, and migration in promoting cancer. Mol
Carcinog. 2022;61(2):173-199.
doi pubmed - Singh RK, Najmi AK, Dastidar SG. Biological functions and
role of mitogen-activated protein kinase activated protein kinase 2 (MK2) in inflammatory
diseases. Pharmacol Rep. 2017;69(4):746-756.
doi pubmed - Alspach E, Flanagan KC, Luo X, Ruhland MK, Huang H, Pazolli
E, Donlin MJ, et al. p38MAPK plays a crucial role in stromal-mediated tumorigenesis. Cancer
Discov. 2014;4(6):716-729.
doi pubmed - Berggren KL, Restrepo Cruz S, Hixon MD, Cowan AT, Keysar SB,
Craig S, James J, et al. MAPKAPK2 (MK2) inhibition mediates radiation-induced inflammatory
cytokine production and tumor growth in head and neck squamous cell carcinoma. Oncogene.
2019;38(48):7329-7341.
doi pubmed - Ray AL, Berggren KL, Restrepo Cruz S, Gan GN, Beswick EJ.
Inhibition of MK2 suppresses IL-1beta, IL-6, and TNF-alpha-dependent colorectal cancer growth.
Int J Cancer. 2018;142(8):1702-1711.
doi pubmed - Yao K, He L, Gan Y, Liu J, Tang J, Long Z, Tan J. HMGN5
promotes IL-6-induced epithelial-mesenchymal transition of bladder cancer by interacting with
Hsp27. Aging (Albany NY). 2020;12(8):7282-7298.
doi pubmed - Kumar B, Koul S, Petersen J, Khandrika L, Hwa JS, Meacham
RB, Wilson S, et al. p38 mitogen-activated protein kinase-driven MAPKAPK2 regulates invasion of
bladder cancer by modulation of MMP-2 and MMP-9 activity. Cancer Res.
2010;70(2):832-841.
doi pubmed - Kumar B, Sinclair J, Khandrika L, Koul S, Wilson S, Koul HK.
Differential effects of MAPKs signaling on the growth of invasive bladder cancer cells.
Int J Oncol. 2009;34(6):1557-1564.
doi pubmed - Manke IA, Nguyen A, Lim D, Stewart MQ, Elia AE, Yaffe MB.
MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase
progression in response to UV irradiation. Mol Cell. 2005;17(1):37-48.
doi pubmed - Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient
cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for
survival after DNA damage. Cancer Cell. 2007;11(2):175-189.
doi pubmed - Sur S, Agrawal DK. Phosphatases and kinases regulating CDC25
activity in the cell cycle: clinical implications of CDC25 overexpression and potential
treatment strategies. Mol Cell Biochem. 2016;416(1-2):33-46.
doi pubmed - Reinhardt HC, Hasskamp P, Schmedding I, Morandell S, van
Vugt MA, Wang X, Linding R, et al. DNA damage activates a spatially distinct late cytoplasmic
cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol Cell.
2010;40(1):34-49.
doi pubmed - Morandell S, Reinhardt HC, Cannell IG, Kim JS, Ruf DM, Mitra
T, Couvillon AD, et al. A reversible gene-targeting strategy identifies synthetic lethal
interactions between MK2 and p53 in the DNA damage response in vivo. Cell Rep.
2013;5(4):868-877.
doi pubmed - Dietlein F, Kalb B, Jokic M, Noll EM, Strong A, Tharun L,
Ozretic L, et al. A Synergistic Interaction between Chk1- and MK2 Inhibitors in KRAS-Mutant
Cancer. Cell. 2015;162(1):146-159.
doi pubmed - Kong YW, Dreaden EC, Morandell S, Zhou W, Dhara SS, Sriram
G, Lam FC, et al. Enhancing chemotherapy response through augmented synthetic lethality by
co-targeting nucleotide excision repair and cell-cycle checkpoints. Nat Commun.
2020;11(1):4124.
doi pubmed - Luo D, Mladenov E, Soni A, Stuschke M, Iliakis G. The
p38/MK2 pathway functions as Chk1-backup downstream of ATM/ATR in G(2)-checkpoint activation in
cells exposed to ionizing radiation. Cells. 2023;12(10).
doi pubmed - Mobley SR, Liu TJ, Hudson JM, Clayman GL. In vitro growth
suppression by adenoviral transduction of p21 and p16 in squamous cell carcinoma of the head and
neck: a research model for combination gene therapy. Arch Otolaryngol Head Neck Surg.
1998;124(1):88-92.
doi pubmed - Yip HT, Chopra R, Chakrabarti R, Veena MS, Ramamurthy B,
Srivatsan ES, Wang MB. Cisplatin-induced growth arrest of head and neck cancer cells correlates
with increased expression of p16 and p53. Arch Otolaryngol Head Neck Surg.
2006;132(3):317-326.
doi pubmed - Cooper MJ, Haluschak JJ, Johnson D, Schwartz S, Morrison LJ,
Lippa M, Hatzivassiliou G, et al. p53 mutations in bladder carcinoma cell lines. Oncol Res.
1994;6(12):569-579.
pubmed - Lemaire M, Froment C, Boutros R, Mondesert O, Nebreda AR,
Monsarrat B, Ducommun B. CDC25B phosphorylation by p38 and MK-2. Cell Cycle.
2006;5(15):1649-1653.
doi pubmed - Beyaert R, Cuenda A, Vanden Berghe W, Plaisance S, Lee JC,
Haegeman G, Cohen P, et al. The p38/RK mitogen-activated protein kinase pathway regulates
interleukin-6 synthesis response to tumor necrosis factor. EMBO J.
1996;15(8):1914-1923.
pubmed - Ronkina N, Menon MB, Schwermann J, Tiedje C, Hitti E,
Kotlyarov A, Gaestel M. MAPKAP kinases MK2 and MK3 in inflammation: complex regulation of TNF
biosynthesis via expression and phosphorylation of tristetraprolin. Biochem Pharmacol.
2010;80(12):1915-1920.
doi pubmed - Mahtani KR, Brook M, Dean JL, Sully G, Saklatvala J, Clark
AR. Mitogen-activated protein kinase p38 controls the expression and posttranslational
modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Mol
Cell Biol. 2001;21(19):6461-6469.
doi pubmed - Clement SL, Scheckel C, Stoecklin G, Lykke-Andersen J.
Phosphorylation of tristetraprolin by MK2 impairs AU-rich element mRNA decay by preventing
deadenylase recruitment. Mol Cell Biol. 2011;31(2):256-266.
doi pubmed - Ryu J, Yoon NA, Lee YK, Jeong JY, Kang S, Seong H, Choi J,
et al. Tristetraprolin inhibits the growth of human glioma cells through downregulation of
urokinase plasminogen activator/urokinase plasminogen activator receptor mRNAs. Mol Cells.
2015;38(2):156-162.
doi pubmed - Ryu J, Yoon NA, Seong H, Jeong JY, Kang S, Park N, Choi J,
et al. Resveratrol Induces Glioma Cell Apoptosis through Activation of Tristetraprolin. Mol
Cells. 2015;38(11):991-997.
doi pubmed - Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA,
Diaz-Latoud C, Gurbuxani S, et al. Hsp27 negatively regulates cell death by interacting with
cytochrome c. Nat Cell Biol. 2000;2(9):645-652.
doi pubmed - Del Rosario O, Suresh K, Kallem M, Singh G, Shah A, Zheng L,
Yun X, et al. MK2 nonenzymatically promotes nuclear translocation of caspase-3 and resultant
apoptosis. Am J Physiol Lung Cell Mol Physiol. 2023;324(5):L700-L711.
doi pubmed - Suresh K, Del Rosario O, Kallem M, Singh G, Shah A, Zheng L,
Yun X, et al. Tumor MK2 transcript levels are associated with improved response to chemotherapy
and patient survival in non-small cell lung cancer. Physiol Genomics.
2023;55(4):168-178.
doi pubmed - Grierson PM, Dodhiawala PB, Cheng Y, Chen TH, Khawar IA, Wei
Q, Zhang D, et al. The MK2/Hsp27 axis is a major survival mechanism for pancreatic ductal
adenocarcinoma under genotoxic stress. Sci Transl Med. 2021;13(622):eabb5445.
doi pubmed
This
article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0
International License, which permits unrestricted non-commercial use, distribution, and
reproduction in any medium, provided the original work is properly cited.
World
Journal of Oncology is published by Elmer Press Inc.